
Executive Summary
U.S. law grants the Food and Drug Administration the power to make consumers get a prescription before purchasing certain drugs. The rationale behind government-imposed prescription requirements is consumer safety—that is, the idea that some drugs are too dangerous for consumers to use without physician supervision.
Research shows, however, that government routinely requires prescriptions for drugs that are safe for consumers to use on their own. For years, Food and Drug Administration (FDA) prescription requirements steered consumers away from safer nonsedating antihistamines toward more dangerous sedating antihistamines. More recently and for political reasons, Presidents George W. Bush and Barack Obama collectively blocked access to “Plan B” emergency contraception for more than 12 years. The FDA continues to force consumers to endure unnecessary and costly visits to their doctors before obtaining routine-use oral contraceptives and life-saving drugs such as naloxone.
Government-imposed prescription requirements violate the rights of individuals to access the medicines they want. Vesting this power in government has left Americans with less access to medicines overall—even relative to consumers in other nations where governments also impose prescription requirements. It imposes unnecessary costs that rise during public health crises such as the COVID-19 pandemic. Evidence also suggests that government-imposed prescription requirements make patients less safe, not more.
Congress should deny the FDA any power to impose prescription requirements. Doing so would not end prescription requirements. The threat of tort liability would push pharmaceutical manufacturers to require authorization from a physician or other competent medical professional before consumers could purchase unusually dangerous drugs. Even without a statutory requirement, consumers would continue to consult health care professionals before accessing certain drugs when they see the need for expert advice. Drug manufacturers, pharmacies, and their liability insurers could develop innovative means of tailoring drug access to the risks that individual drugs pose.
Denying government the power to require prescriptions would expand drug access by reducing both drug prices and the associated nonprice costs of obtaining needed drugs. The evidence suggests that eliminating government-imposed prescription requirements would lead to more-judicious use of pharmaceuticals because consumers make more-cautious drug decisions when the choice is theirs rather than when government forces them to consult physicians. Denying the FDA this power would help ensure access to beneficial medicines during the COVID-19 pandemic and subsequent public health crises.
Introduction
The United States leads the world in per capita spending on pharmaceuticals. Inflation-adjusted per capita spending on retail prescription drugs grew from $90 in 1960 to $1,025 in 2017.1 Spending on pharmaceuticals constituted 12 percent of overall health care spending in 2017.2
An important contributor to relatively high drug spending in the United States is relatively high drug prices. One study that examined high-income countries from 2013 to 2016 found:
Among the 11 countries, the United States had the highest pharmaceutical spending per capita at $1,443, with Switzerland following at $939 and a mean of $749.… For 4 pharmaceuticals (Crestor, Lantus, Advair, and Humira) used for common conditions, the United States had higher prices than all other countries; for 3 of these, the U.S. price was more than double the next highest price.…
The United States spent approximately twice as much as other high-income countries on medical care, yet utilization rates in the United States were largely similar to those in other nations. Prices of labor and goods, including pharmaceuticals, and administrative costs appeared to be the major drivers of the difference in overall cost between the United States and other high-income countries.3
A 2019 study found that of the 36 top-selling drugs on the market in the United States from 2012 through 2017, “28 (78%) have seen an increase in insurer and out-of-pocket costs by more than 50%, and 16 (44%) have more than doubled in price.”4
President Trump has claimed that pharmaceutical manufacturers are “getting away with murder.” His administration has unveiled several minor proposals mainly aimed at increasing price competition and accelerating the approval of generic drugs.5 While somewhat palliative, the proposals would leave the United States’ costly pharmaceutical regulatory regime fundamentally unaltered.6 Improving consumer access to pharmaceuticals requires fundamentally rethinking pharmaceutical regulation.

The current U.S. pharmaceutical regulatory regime is paternalistic. It restricts autonomy by denying consumers the right to self-medicate and sometimes denies desperate patients an opportunity to save their own lives. It places the judgment of perceived experts above the autonomy of the individual. It fails to appreciate, much less replicate, the dynamism and responsiveness of a free market. It inhibits new drug development and contributes to excessive drug prices.
Removing the paternalistic features of drug regulation would promote greater choice, innovation, and affordability while restoring respect for the dignity and autonomy of the individual. Federal law grants the U.S. Food and Drug Administration (FDA) the power to prevent competent adults from accessing certain drugs unless they spend time and money to get a government-mandated permission slip (i.e., a prescription). The FDA interpreted the Food, Drug, and Cosmetic Act of 1938 (FDCA) as granting it this power, even though the bill’s sponsors disavowed any such desire. Congress codified this usurped power in the 1951 Durham-Humphrey Amendment to the FDCA. Neither step was responsive to any contemporaneous drug crisis or regulatory failure.
The rationale behind government-imposed prescription requirements is consumer safety (i.e., that some drugs are too dangerous for consumers to use without physician supervision). It may be comforting to imagine this power only finds its way into the hands of wise policymakers whose sole motivation is a selfless concern for consumers; the reality is not quite so idyllic.
Researchers find that governments routinely use prescription requirements to restrict access to drugs that are safe for consumers to use on their own. For years, the FDA let consumers purchase cheap, sedating antihistamines without prescriptions but required prescriptions for nonsedating antihistamines, which are less dangerous. More recently, Presidents George W. Bush and Barack Obama blocked access to “Plan B” emergency contraception for a combined 12 years for political reasons. The FDA continues to force consumers to endure unnecessary and costly visits to doctors before obtaining routine-use oral contraceptives (commonly called “the pill”) and life-saving drugs such as naloxone.
Government-imposed prescription requirements violate individual rights and increase the cost of accessing beneficial drugs. They force consumers to undertake the time and expense of seeing a physician—an expense that is also higher in the United States than in other advanced nations—and may even contribute to higher prices for pharmaceuticals that consumers could otherwise afford and use safely on their own. Vesting this power in government has left Americans with less access to medicines than consumers in many other nations. It also has imposed unnecessary costs that rise during public health crises such as the COVID-19 pandemic. Evidence suggests government-imposed prescription requirements make consumers less safe, not more.
Congress should repeal Durham-Humphrey and amend federal law to deny the FDA any power to impose prescription requirements. This would not end prescription requirements. The threat of tort liability would lead pharmaceutical manufacturers to require authorization from a physician or other competent medical professional before consumers could purchase unusually dangerous drugs. Pharmaceutical companies could also tailor drug access to the risks that individual drugs pose, such as by designating drugs to be available for sale on a pharmacist-only, behind-the-counter, over-the-counter, or other basis. To ensure reform restores the right of individuals to self-medicate, Congress should also eliminate premarket approval requirements for new drugs.
The best evidence suggests that eliminating government-imposed prescription requirements would lead to more-judicious use of pharmaceuticals because consumers make more-cautious drug decisions when the choice is theirs rather than when government forces them to consult physicians. Even without a statutory requirement, consumers would continue to consult health care professionals before accessing certain drugs when they see the need for expert advice. Repeal would help restore individual autonomy and dignity while reducing drug prices and the associated nonprice costs of accessing beneficial drugs.
The Right to Self-Medicate
The notion that competent adults have the right to self-medicate is a corollary of the doctrine of informed consent. The doctrine asserts that individuals have a right to refuse whatever medical treatment they choose, even if doing so will harm them. The right to self-medicate is the idea that individuals likewise have a right to use whatever medical treatments they choose, even if doing so will harm them.
Patients-rights advocates struggled for years to force the medical profession to respect patient autonomy through the doctrine of informed consent. Traditionally, an ethos of medical paternalism governed relationships between physicians and patients. Doctors routinely deceived or withheld information from patients about the patients’ health based on the physicians’ judgment of the patients’ interests. In some cases, doctors would perform invasive procedures on competent adults without their consent—indeed, against patients’ express wishes.
An infamous instance of medical paternalism occurred in 1908, after an elocutionist from San Francisco named Mary Schloendorff consented to let doctors at New York Hospital put her under anesthesia for the purposes of an examination. Schloendorff expressly and repeatedly told her doctors and nurses that she did not consent to any operation. Her physicians nevertheless removed a uterine fibroid tumor while she was unconscious. Schloendorff blamed the unwanted procedure for the subsequent gangrene in her left arm and the resulting amputation of multiple fingers. She sued the hospital for the tort of assault.7
In Schloendorff v. Society of New York Hospital, New York Court of Appeals Judge Benjamin Cardozo’s opinion became the basis of the doctrine of informed consent and present-day medical ethics:
In the case at hand, the wrong complained of is not merely negligence. It is trespass. Every human being of adult years and sound mind has a right to determine what shall be done with his own body; and a surgeon who performs an operation without his patient’s consent, commits an assault, for which he is liable in damages. This is true except in cases of emergency where the patient is unconscious and where it is necessary to operate before consent can be obtained.… [Schloendorff] had never consented to become a patient for any purpose other than an examination under ether.… She had forbidden the operation.8
The court determined that Schloendorff’s doctors had committed an assault. (It nevertheless ruled against her because she had sued the hospital, which the court found not liable for the actions of the doctors who practiced there.)
For decades, the medical profession resisted the doctrine of informed consent and the underlying goal of medical autonomy. Even into the 1970s, many doctors admitted to withholding terminal cancer diagnoses from their patients.9
Today, the patient-doctor relationship has largely shifted from one of medical paternalism and patient acquiescence to what bioethicist Daniel Sokol calls a “leveled partnership” in which the medical profession respects patient autonomy and the government punishes providers who violate the doctrine of informed consent.10
A notable exception to the new ethos of individual autonomy exists in access to pharmaceuticals. Bioethicist Jessica Flanigan argues that government-mandated premarket approval and prescription requirements are forms of coercive medical paternalism that interfere with individual autonomy as much as when doctors lie to patients about the patients’ diagnoses, prognoses, or treatment options or perform unauthorized procedures on them:
Paternalism is just as wrong at the pharmacy as it is in the doctor’s office. Medical autonomy is an important value in both contexts, so states should protect patients’ rights against unwanted medical interventions from physicians and from unwanted limits on access by public officials. Both informed consent requirements and rights of self-medication will permit people to make decisions that their physicians would advise against.11
Flanigan identifies two areas in which government may ethically restrict the ability to self-medicate. One is antibiotics. Consumers who use antibiotics indiscriminately promote the development of antibiotic-resistant organisms, which can potentially expose others to risk of harm or even death from infectious diseases.12 It is therefore ethically permissible, Flanigan argues, for government to restrict the use of certain antibiotics. The other area pertains to children and to adults with severe cognitive disabilities who have autonomous capacities not dissimilar to children. Such individuals are unable to make medical decisions in accordance with the doctrine of informed consent and therefore cannot claim the right to self-medication. Flanigan argues that it is therefore ethically permissible to restrict their access to medications.13
The doctrine of informed consent and the right to self-medicate are inextricably linked. Any argument that individuals do not have a right to self-medicate necessarily undermines the doctrine of informed consent. It is impossible to infringe on one without threatening the other. If one is valid, so is the other. If one supports the doctrine of informed consent, one must logically respect the right to self-medicate.
A Brief History of U.S. Pharmaceutical Regulation
The history of pharmaceutical regulation in the United States shows that the public and Congress traditionally respected the right to self-medicate. In 1938, however, the FDA without justification effectively began imposing prescription requirements by fiat, claiming it could do so under the FDCA. In 1951, Congress endorsed and codified this power grab. As consumer advocates were struggling to defend medical-autonomy rights through adoption of the doctrine of informed consent, the U.S. government turned against individual autonomy and codified legal and medical paternalism in the area of pharmaceuticals by allowing government officials and medical practitioners to substitute their values and judgment for those of consumers. Access to pharmaceuticals remains stuck in a model in which the relationship between doctors and consumers resembles that of guardians and their wards.
Pre-1938
Prior to 1938, federal law generally respected the right of individuals to self-medicate. With narrow exceptions such as the Harrison Narcotics Act of 1914, which required prescriptions for narcotics that exceeded allowable limits, there were no federal requirements that consumers obtain a prescription from a physician before purchasing a drug. “The status of a drug as prescription or nonprescription was left entirely to the manufacturer,” noted former FDA general counsel Peter Barton Hutt.14 The FDA’s official historian, John P. Swann, wrote:
Manufacturers … had marketed (if not labelled) selected products such as insulin with the intention that they be used only under a physician’s supervision. In fact, as far back as the 1880s, a New York physician related his experience with detail men who assured him that their companies’ preparations were sold only with prescriptions.15
Unless the manufacturer required a prescription, adults were free to purchase any nonnarcotic drug for self-medication without a doctor’s permission.
Nevertheless, economist Sam Peltzman reports that by 1938, “About one-third of drug purchases were being made under a doctor’s prescription.”16 In other words, even when there was no law requiring them to do so, consumers routinely sought expert advice from doctors and dentists. Consumers weighed their physicians’ recommendations alongside other information, including the insights and recommendations of pharmacists. But in the end, consumers themselves decided what advice to follow and what medications to use.
The private sector developed resources to help consumers and their physicians make these decisions. In 1820, voluntary cooperation among physicians, pharmacists, and schools of pharmacy led to the creation of the United States Pharmacopeial Convention, a private, nonprofit organization that exists to disseminate information about pharmaceuticals.17 The organization continues to publish and regularly update the United States Pharmacopeia (USP), an authoritative compendium of drugs and drug uses, including indications, dosage recommendations, warnings, contraindications, and off‐label uses. The organization also produces the National Formulary, a compendium of drugs, dietary supplements, vitamins, and minerals. The National Formulary established standards for composition, purity, strength, storage, and labeling and defines the analytical tests and methods that measure adherence to its standards.18
When the federal government began regulating pharmaceuticals in the early 20th century, it continued to respect the right to self-medicate. In reaction to highly publicized instances of drug manufacturers defrauding, misleading, or even harming consumers, Congress passed the Pure Food and Drugs Act (PFDA) of 1906. Rather than infringe on the right to self-medicate or limit medical autonomy, the PFDA attempted to provide more information to consumers and physicians. The law codified the privately created USP and defined a drug as “adulterated” if it failed to meet the USP’s standards. Those provisions had little apparent effect, as the USP was already the widely recognized standard of practice. The PFDA also defined the crime of “misbranding,” stating that a drug was misbranded if it contained alcohol, opium, cocaine, or any other dangerous or potentially addictive substance and failed to list those ingredients (and their proportional inclusion) on the product label. The U.S. Bureau of Chemistry, which implemented the new law, had no authority to determine the efficacy of pharmaceuticals.19

Congress passed the Pure Food and Drugs Act of 1906 in reaction to highly publicized instances of drug manufacturers defrauding, misleading, or harming consumers. Source: U.S. Capitol Visitor Center.
Other minor changes soon followed. In 1912, the Sherley Amendment allowed prosecution of manufacturers who knowingly made false or fraudulent claims about a drug. In 1927, Congress reorganized the Bureau of Chemistry into the Food, Drug, and Insecticide Administration. In 1930, Congress renamed the agency the Food and Drug Administration (FDA).20
The Food, Drug, and Cosmetics Act of 1938
A highly publicized tragedy spurred major new legislation in 1938. The S. E. Massengill Company had been successfully marketing a safe and effective new antimicrobial called sulfanilamide. The company decided to release a sweet-flavored liquid “elixir” formulation to make the drug easier for children and others to ingest. It produced the sulfa drug according to specifications, but the solvent it used did not meet the USP standard for branding as an elixir. The USP, and therefore the PFDA, allowed only solutions that used alcohol as the solvent to bear the name “elixir.” Rather than alcohol, the company used diethylene glycol—a close chemical cousin of antifreeze (ethylene glycol). The solution poisoned hundreds of consumers, causing extremely painful reactions and 105 deaths, including 34 children.21

Six‐year‐old Jo Anne Cramer of Ghent, Ohio, died after she ingested elixir sulfanilamide.
Under pressure from the FDA, S. E. Massengill recalled the product. The FDA fined the company $26,100—“the highest that was legally allowed at the time”—but not for killing people or even for failing to conduct safety testing.22 The PFDA only allowed the FDA to fine the company for mislabeling the drug an elixir. “If the product had been called a ‘solution’ instead of an ‘elixir,’ no charge of violating the law could have been made.”23 S. E. Massengill settled suits out of court with family members of elixir sulfanilamide victims.24 The chemist responsible committed suicide.
In response to this tragedy, Congress passed the FDCA. Among its key provisions was a requirement that manufacturers file a new drug application with the FDA before they could market any new drug. The application had to include information on the drug’s composition, safety test results, and the manufacturer’s quality controls. If the FDA approved the drug as safe, or failed to act on it within 60 days, the manufacturer could proceed to market. (The FDCA allowed existing drugs with a record of proven safety to remain on the market.) The FDCA also imposed stricter misbranding rules. It required manufacturers to list all ingredients in their precise amounts on labels. Crucially, it imposed expensive new labeling requirements for all drugs—and created the potential for the FDA to issue exemptions from those requirements.
The FDCA did not explicitly require manufacturers to designate any drugs as prescription-only.25 Indeed, even as Congress debated and enacted the FDCA, its supporters paid homage to the right to self-medicate. Supporters argued that the FDCA was merely a truth-in-labeling bill that sought to make self-medication safer by furnishing consumers more information. In testimony to Congress in support of an early iteration of the law, FDA chief Walter G. Campbell repeatedly affirmed that the bill’s purpose and effect would be to facilitate self-medication, not restrict it:
There is no issue, as I have told you previously, from the standpoint of the enforcement of the Food and Drugs Act about self-medication. This bill does not contemplate its prevention at all.… But what is desired … is to make self-medication safe.
[The bill provides] information that will permit the intelligent and safe use of drugs for self-medication.… All of the provisions dealing with drugs, aside from those recognized in the official compendia, are directed towards safeguarding the consumer who is attempting to administer to himself. If this measure passes, self-medication will become infinitely more safe than it has ever been in the past.26
Sen. Royal S. Copeland (D‑NY), who was a homeopathic physician, said of an early version of the bill:
There is no more common or mistaken criticism of this bill than that it denies the right to self-medication.… Nothing could be further from the truth. The proposed law simply contributes to the safety of self-medication by preventing medicines from being sold as “cures” unless they really are cures. It requires that drugs which have only palliative effect say as much on the label.27
The House committee that reported the bill in 1938 wrote: “The bill is not intended to restrict in any way the availability of drugs for self-medication. On the contrary, it is intended to make self-medication safer and more effective.”28
This only made sense. The elixir sulfanilamide tragedy provided no justification for curtailing the right to self-medicate. A government-imposed prescription requirement would not have prevented the tragedy; of the 105 consumers who died, 100 took the drug under the direction of government-licensed physicians.29 Nor have there since been any comparable drug poisoning tragedies for which self-medication was the culprit. If anything, the elixir sulfanilamide tragedy offers evidence that, even in the absence of government-imposed prescription requirements, consumers overwhelmingly seek advice from trusted experts before taking medications—even if those experts sometimes do not deserve their patients’ trust. (See discussions of thalidomide in the “Another Drugmaker Kills; Government Again Restricts Consumer Rights” section and of elixir sulfanilamide and thalidomide in the “How Could Consumer Safety Regulation Make Consumers Less Safe?” section).
Despite the assurances of its supporters, the FDCA nevertheless facilitated the system of federally mandated prescription requirements that exists today. The act’s labeling requirements stated:
A drug or device shall be deemed to be misbranded … (f) Unless its labeling bears (1) adequate directions for use; and (2) such warnings against use in those pathological conditions or by children where its use may be dangerous to health, or against unsafe dosage or methods or duration of administration or application, in such manner and form, as the Secretary finds necessary for the protection of users and by regulation prescribes: Provided, That where any requirement of clause (1) of this paragraph, as applied to any drug or device, is not necessary for the protection of the public health, the Secretary shall promulgate regulations exempting such drug or device from such requirement.30
This section required drug labels to bear instructions for use and to list effects and possible side effects in a manner that a person with little education could understand.31
Manufacturers expressed several concerns about these labeling requirements. For starters, compliance was expensive. One drug industry representative complained:
Every one of these labels have got to be changed. It is a fearful job. They have to print them by the millions. Many of these products sell 8, 10, 20 million packages and more a year. They have to be bought months in advance, and go into production months in advance. Millions of dollars are involved not only in discarding stocks of goods on hand, mind you, but in the purchase of these tens of millions of new labels.32
Manufacturers further complained that some drugs were so dangerous or that safe administration was so complex that it would be impossible to write a label with “adequate directions for use” that would be intelligible to laypeople. One manufacturer wrote:
Under the proposed regulation the labeling must include a full and complete description of the conditions, with their symptoms, for which the preparation is indicated, and a statement of the treatment thereof in such detail that every consumer may determine the proper course of self-medication. In effect, a correspondence course in medicine is to be afforded to laymen.…
The warning that phenobarbital is contraindicated in large doses in nephritic subjects is ineffectual and meaningless to a lay consumer who does not know that he is suffering from nephritis (even if the labeling use the synonym “Bright’s disease”). Furthermore, a layman lacks the knowledge and experience to determine what quantity constitutes a large dose or an excessive amount. In some cases a layman may not recognize the presence of untoward effects specified in the warning until considerable harm has resulted from continued use of the drug.33
In other words, manufacturers of certain dangerous drugs were taking steps to protect consumers from them by labeling such drugs for, and marketing them exclusively to, physicians who would then prescribe them to patients. The FDCA exposed consumers to greater harm by requiring such manufacturers to affix to those drugs a label with instructions for lay use.
Manufacturers reasonably argued that forcing them to label such drugs for consumer use could make consumers less safe by misleading them to believe that they could use the drugs without physician supervision and could thereby expose manufacturers to liability.34
At the behest of manufacturers, the FDA promulgated regulations exempting drugs from the new labeling requirements if the manufacturer marketed the drugs solely to physicians and solely on a prescription-only basis.35 A label qualified for the exemption, the FDA ruled, if it bore the warning, “Caution: To be used only by or on the prescription of a physician.”36
The combination of a costly and coercive labeling requirement alongside an exemption for prescription-only drugs predictably transformed a law that purported to facilitate self-medication into a sweeping curtailment of the right to self-medicate. The expense and liability associated with compliance with the labeling requirement effectively coerced manufacturers into selling many drugs on a prescription-only basis. Swann wrote, “Manufacturers began … labelling many drugs that were safe for self-medication with the prescription legend.”37 According to Massachusetts Institute of Technology economic historian Peter Temin: “The drug firms introduced virtually all of the new drugs as prescription drugs, apparently without sustained opposition. Since drug firms are engaged in profit-making activity, they undoubtedly thought they derived a commercial advantage from this designation.”38
Commercial advantage and risk aversion pushed in the same direction. Classifying or reclassifying drugs as prescription-only was likely attractive to drug manufacturers because it allowed them to charge higher prices (see the “A Contributor to Excessive Drug Prices” section), sidestep extremely costly labeling requirements, and avoid enforcement actions by an agency that provided almost no guidance as to how to proceed. In 1982, Hutt wrote that the “general principles” the agency offered about how to classify drugs were “so vague and indeterminate as to provide virtually no guidance for daily decisions.” The FDA “never enunciated either in published regulations or in other written documents the kind of operational rules that would provide clear policy and result in consistent decisions on the prescription/nonprescription status of drugs.”39 In other words, the FDA restricted the right to self-medicate through both action and inaction.
The FDCA’s implementation thus belied the assurances of the law’s authors. As Peltzman put it: “A law that was written (ostensibly) to promote more informed choice by consumers was interpreted instead to restrict consumer choice.”40 Temin summarized this “stunning” and unauthorized usurpation of the consumer’s right to choose:
The Federal Food, Drug, and Cosmetic Act of 1938 … undertook to assure the public that any drug on the market could be taken in reasonable quantities without harm. The government thereby restricted the range of consumer choice by taking harmful substances off the market. But the layman was still free to choose his own drugs from among all nonharmful, nonnarcotic drugs. He could consult a doctor if he wished, but he was under no obligation to do so.
By the end of 1938, the FDA had announced that the government would sharply curtail this freedom of choice. Consumers, the FDA said, were not competent to make their own drug choices.… The government had delegated the consumers’ choice to manufacturers and doctors—and nobody commented.…
This change in the underlying assumptions of drug legislation came about through internal FDA processes. The shift from assuming a capable consumer to assuming an incompetent consumer was made within the FDA within six months of the Federal Food, Drug, and Cosmetics Act’s passage. Not only was the shift in assumptions not controversial, the method by which it was accomplished occasioned no comment as well. The decisions of the FDA were ratified by the courts and enacted into statute by the Congress. Neither branch of the government undertook to question the FDA’s assumptions.41
Even if the FDCA’s authors had intended all along to restrict the right to self-medicate, their denials that the act would do so show that this right enjoyed broad support before the law’s enactment. The FDA’s frustration that consumers kept purchasing many prescription-only drugs without prescriptions, and the agency’s many prosecutions of pharmacists for selling such drugs without prescriptions, further illustrate that the right to self-medicate enjoyed broad support even after the law’s enactment. Such civil disobedience continued for at least a decade.42
The Durham-Humphrey Amendment of 1951
Official respect for the right to self-medicate ended in 1951 with the passage of the Durham-Humphrey Amendment. Despite large incentives to designate drugs as prescription-only, many drug manufacturers continued to sell drugs directly to consumers. The lack of guidance from the FDA led some manufacturers to require prescriptions for drugs for which other manufacturers did not.43 The situation created confusion and fear of prosecution among pharmacists.44 With the support of their profession, two former pharmacists—Rep. Carl Durham (D‑NC) and Sen. Hubert Humphrey (D‑MN)—sponsored an amendment to the FDCA that they argued would bring uniformity to the market for prescription drugs.45
The Durham-Humphrey Amendment formally authorized the FDA to classify pharmaceuticals as either over-the-counter (OTC) or prescription-only. The amendment prohibits dispensing a prescription-only drug unless the consumer presents a prescription from a government-licensed health care practitioner. Manufacturers can request OTC or prescription-only classification when submitting a new drug application, but the FDA makes the ultimate decision. The amendment exempted drugs already on the market that had a proven safety record, which is why a few of the original brands of insulin extracted from agricultural animals and already on the market prior to the 1938 FDCA (e.g., Humulin and Novo-Novolin) are still available in most states without a prescription.46 The Durham-Humphrey Amendment further established rules and procedures for switching a drug’s classification from prescription-only to OTC, allowed for prescriptions with authorized refills (prescriptions had previously been single-use only), and allowed for doctors to phone in prescriptions if pharmacies immediately converted them to writing.47
Whereas the FDA’s interpretation of the FDCA implicitly coerced manufacturers into marketing many drugs as prescription-only, Durham-Humphrey explicitly denied drug manufacturers the right to decide whether consumers must consult with a clinician before accessing a drug and the right to decide from what type of clinician consumers must obtain a prescription. It therefore also gave the FDA the power to deny consumers the right to choose whether to self-medicate or to medicate under the direction of a clinician. The FDA’s interpretation of the FDCA and the Durham-Humphrey Amendment thus increased the cost of obtaining many drugs by requiring consumers to spend more time and money getting prescriptions. The agency’s continued failure to provide clear guidance about how to classify drugs again created incentives for risk-averse manufacturers to request prescription-only status, even for safe drugs, to reduce the risk of FDA enforcement actions.48
Durham-Humphrey does leave states some power to help their residents—the power to decide which health care practitioners may write prescriptions. States can therefore expand access to medications by letting nonphysician clinicians—pharmacists, nurse practitioners, etc.—prescribe drugs. Some states have used this strategy broadly or for specific drugs.
This aspect of the amendment is a double-edged sword, however. By increasing the number of drugs for which consumers must obtain prescriptions, Durham-Humphrey also increases economic incentives for physicians to lobby state legislatures against expanding prescriptive authority to other clinicians.
Another Drugmaker Kills; Government Again Restricts Consumer Rights
In the years that followed, Congress continued to increase federal regulation of pharmaceuticals. Among the most significant changes were the Kefauver-Harris Amendments of 1962. These amendments exacerbated the effects of the Durham-Humphrey Amendment’s government-imposed prescription requirements.
Like the FDCA that they amended, the Kefauver-Harris Amendments were a response to a tragedy involving unsafe drugs. In recent decades, the FDA has approved the drug thalidomide to treat leprosy and multiple myeloma.49 The drug’s adverse side effects, however, include severe and often fatal birth defects when pregnant women take it.50 Humanity learned this the hard way: when manufacturers marketed thalidomide as a sedative in the late 1950s and early 1960s, it led to an estimated 10,000 cases of fetal abnormalities across 46 countries.51 Many fetuses died in utero or shortly after birth.
Thalidomide affected relatively few Americans because the FDA, citing safety concerns, refused to approve the drug. “Nonetheless,” writes physician and historian Paul M. Wax, “under the guise of conducting a new-drug investigation, the Merrell Pharmaceutical Company managed to supply more than 2,500,000 thalidomide tablets to 1,270 physicians in the United States for investigational use.”52 Those government-licensed physicians then prescribed the drug to 20,771 patients, including 3,879 women of child-bearing age, 624 of whom were pregnant.53 These physicians apparently did not tell their patients that the drug was experimental or that the FDA had not approved it.54 The FDA reported that there were “17 children born in America with thalidomide-associated deformities.”55
Spurred to action by news reports and horrifying photos of “thalidomide babies,” Congress passed the Kefauver-Harris Amendments. The amendments require drug companies to conduct additional tests and trials to demonstrate that new drugs are safe. For the first time, Congress also required manufacturers to establish to the FDA’s satisfaction that a new drug is effective at treating a specific condition. The amendments require “that informed consent be obtained from all research study subjects so that patients would have to be specifically informed if a drug they were being given or prescribed was ‘experimental,’ something that had not happened in the case of thalidomide.”56 The amendments also eliminated requirements that the FDA approve or deny a new drug application within a specified period. Wax explains that with the Kefauver-Harris Amendments, “the transition of the FDA from an agency responding to events to an agency actively scrutinizing new-drug development was complete.”57

A letter from the Wm. S. Merrell Company advises of the possible relationship between thalidomide and birth defects.
Imposing a proof-of-efficacy requirement before manufacturers can take a new drug to market was a curious response to the thalidomide tragedy. The concern with thalidomide was not its efficacy. It is indeed effective as a sedative—and in treating nausea associated with pregnancy for that matter. The issue with thalidomide was safety, and the FDA already had sufficient authority to keep it off the market until it was proven safe, as the agency’s handling of the drug demonstrates. One can nevertheless argue for a proof-of-efficacy requirement on safety grounds. Establishing efficacy requires manufacturers to conduct more and longer clinical trials that often identify adverse drug reactions that smaller, safety-focused trials do not.
What makes the proof-of-efficacy requirement most curious is that Congress’s response to malfeasance on the part of pharmaceutical companies and doctors was to punish patients by further curtailing their freedom to self-medicate. The Durham-Humphrey Amendment interferes with consumers’ freedom to self-medicate by requiring them to get permission from a government-anointed gatekeeper before purchasing certain drugs. The Kefauver-Harris Amendments interfere with consumers’ freedom to self-medicate by delaying for years consumers’ ability either to exercise that right (in the case of OTC drugs) or even to medicate with a physician’s permission (in the case of prescription drugs).
Kefauver-Harris created the twin problems of “drug lag” and “drug loss.” Drug lag refers to the additional time the FDA’s proof-of-efficacy requirement forces consumers to wait before they may access a drug. Every day that the FDA adds to the drug development and approval process is a day that the agency denies consumers their right to self-medicate with that drug. In 1994, economists David Dranove and David Meltzer noted the effects of Kefauver-Harris on total drug development time:
Since the Food and Drug Administration (FDA) Amendments of 1962, the average time from a drug’s first worldwide patent application to its approval by the FDA has risen from 3.5 to 13.5 years.…
Our results indicate that, beginning in the 1950s, more important drugs—especially drugs that proved to be successful in the marketplace—have been developed and approved more rapidly than less important drugs. Despite this, the overall trend of increasing average development and approval times implies that even drugs two standard deviations above the mean level of importance are taking longer and longer to reach the market.58
Even important drugs saw median time to market grow by 7.5 years. The authors cited “several strands of evidence that suggest that the acceleration is due more to the actions of firms than to the FDA initiatives of the mid-1970s” and that “the accelerated approval of important drugs was a worldwide phenomenon.”59 Other studies have found that the time required to bring a drug through FDA-mandated clinical testing and marketing approval alone rose from 7.5 years (90.3 months) in the 1980s and 1990s to 8 years (96.3 months) in the 1990s and 2000s.60 These estimates do not count the preclinical phase of drug development, between synthesis of a new chemical entity and human testing, which adds several years to the FDA approval process. Drug lag is cruelest to terminally ill patients, whom it denies the right to try to save their lives by using a drug already proven safe but awaiting efficacy approval. Many seriously ill Americans die waiting for the FDA to approve drugs that regulators in other countries have already approved.61
Drug loss occurs when pharmaceutical manufacturers choose not to invest in finding new treatments that they do not believe can recoup the considerable cost of securing FDA approval. Presented in 2019 dollars, the average estimated cost of each new drug approval has risen from $523 million in 1987 to $1.2–1.8 billion in 2000 to $3.2 billion in 2013. The cost grew at an average annual real rate of 9.4 percent in the 1970s, 7.4 percent in the 1980s, and 8.5 percent from 1990 through the early 2010s.62 Drug loss denies consumers the freedom to access drugs that would have a lower expected benefit-to-cost ratio than the FDA might accept, or whose potential market is too small to recoup the cost of an FDA approval. The fact that drug loss denies manufacturers’ freedom to bring those drugs to market makes it no less an infringement on consumers’ right to self-medicate.
The high cost of the FDA’s approval processes necessarily leads to higher drug prices. It also creates incentives for manufacturers to market drugs as prescription-only so that they can charge higher prices to help recoup those costs. (See the “A Contributor to Excessive Drug Prices” section.)
Dissatisfaction with the length of the FDA’s approval process led to a national “Right to Try” movement that spurred legislation at the state level and a federal “Right to Try” law in 2018. These laws allow some terminally ill patients to access drugs that the FDA is blocking from the market.63

A snapshot of the national Right to Try campaign website.
Rather than save lives, the Kefauver-Harris Amendments may cost lives. Keeping new drugs off the market until manufacturers conduct more and larger clinical trials no doubt saves lives by preventing unsafe drugs from coming to market. Yet it also causes patients to suffer and even die while waiting for treatments to clear the FDA’s approval process.
Several studies have estimated that the FDA would save more lives if it reduced the length of its new drug approval process. In 1973, Peltzman studied the reduction in new drug introductions since Kefauver-Harris and concluded:
The main finding is that benefits forgone on effective new drugs exceed greatly the waste avoided on ineffective drugs.…
The estimates imply that the magnitude of the problem of ineffective new drugs prior to 1962 was trivial or that the ability of FDA regulation to reduce the problem is small. At the same time, the reduced flow of new drugs due to the amendments is imposing net losses on consumers which are the rough equivalent of a 5–10 percent excise tax on all prescriptions sold.…
The penalties imposed by the marketplace on sellers of ineffective drugs prior to 1962 seem to have been enough of a deterrent to have left little room for improvement by a regulatory agency.64
Peltzman later elaborated: “If our estimates of the gains and losses from exceptionally beneficial and unsafe drugs, respectively, are at all reasonable, there was already a costly bias in the pre-1962 proof-of-safety requirement.… The risk-return tradeoff was already biased against drug consumers in 1962. The [Kefauver-Harris] amendments have simply exaggerated the bias.”65
A 2004 study by Mary K. Olson examined the effects of accelerated reviews for therapeutically novel drugs. Olson’s estimates indicate that the FDA’s drug approval process poses such a high barrier to entry into the market that reducing the duration and cost of that process (i.e., regulating less) would improve overall health. She writes:
When … health benefits are considered, the evidence suggests that [adverse drug reaction] deaths reduce the net longevity benefits of new drugs by approximately 8% in their first two years on the market. Based on this estimate, the evidence suggests that overall regulators [sic] efforts to speed patient access to novel drugs produces a net gain for society.66
In other words, the health improvements that result from shortening the FDA’s approval process exceed the health losses by a factor of 12 in the first two years. Olson notes several caveats, including that consumers may underreport adverse reactions or that adverse reactions could occur beyond two years. She estimates, however, that even if consumers and physicians fail to report 30 percent of adverse drug reactions, the health losses would still account for just 11 percent of the health gains from bringing beneficial new drugs to market faster. That is, the health gains would still be nine times greater than the health losses.67
A 2006 study examined the effects of the Prescription Drug User Fee Acts of 1992, 1997, and 2002 (PDUFA), which accelerated the FDA’s reviews of new drug applications. Tomas J. Philipson and colleagues estimated the health effects of those faster reviews. They conclude:
Our major findings are that the proportion and timing of withdrawal of drugs approved pre- and post-PDUFA do not differ in a statistically significant way; about 2–3% of approved drugs are withdrawn at the same speed before and after the Acts. In addition, we compute an extreme upper bound on the adverse safety effects induced by PDUFA by assuming that all [new molecular entity] withdrawals after 1992 were due to PDUFA and that there were no benefits associated with the drugs so that their social surplus is measured by the harmful health effects the withdrawn drugs imposed. Using this extreme upper bound on the adverse safety effects of PDUFA, we find that the drugs approved and withdrawn during PDUFA cost about 56,000 life years as compared to the gains in health implicit in the greater speed generated by PDUFA, which are estimated at the equivalent of 180,000 to 310,000 life years.68
Even under unreasonably conservative assumptions, the study estimates that the health benefits that resulted from reducing the barriers to entry into the pharmaceutical market were between three and six times greater than the health losses. The authors then suggested that under more reasonable assumptions, the health benefit-to-cost ratio might be, at the margin, infinite:
By the most plausible measure, the act did not, in fact, have any effect on drug safety: neither the proportion of drugs eventually withdrawn (2 to 3 percent), nor the speed with which they were withdrawn, changed in any statistically significant way since the law’s passage.…
By one interpretation, the analysis suggests there was no trade-off between safety and speed: the increased speed in reviewing applications had no measurable impact on the quality of the review process. But even if there was a price—that is, if hanging on to review procedures before 1992 would have reduced errors that led to deaths—there are very good reasons to believe that the price was worth paying. Faster access to new drugs saved more lives than the release of dangerous drugs could possibly have claimed.69
These results suggest, again, that the FDA’s approval processes are so burdensome that, at the margin, they are harming health rather than improving health and that lowering the barriers to new drugs would improve overall health.
The Olson and Philipson studies examined only the effects of drug lag. To the extent that the FDA’s drug approval process also leads to drug loss, the benefits of reducing the time and financial costs of that process would be even greater.
Blocking Access to Safe, Necessary Drugs
Having a government agency make consumers get prescriptions before they access drugs has surface appeal. Many drugs are dangerous. In many cases, the benefits of having a gatekeeper regulate access will exceed the costs. Allowing government to decide which drugs require gatekeepers and who shall be the gatekeepers, however, inevitably harms consumers by denying them access to beneficial drugs for which either no gatekeeper requirement or only a less-costly gatekeeper is necessary. Indeed, the evidence suggests that government-imposed prescription requirements do more harm than good.
Government-imposed prescription requirements increase the financial and time costs of obtaining beneficial medicines and block access to safe, effective, and critical drugs—often for decades. For example, empowering government to choose which drugs require prescriptions likely increased the cost to American consumers of two common medications, the nonsteroidal anti-inflammatory drugs (NSAIDs) ibuprofen and naproxen, for a decade or more:
- Developers of ibuprofen, which relieves pain and reduces fevers, secured a patent for it in 1961. The FDA approved ibuprofen for prescription-only sale in 1974. The agency did not remove the prescription requirement until 1984.70
- The FDA approved the pain reliever naproxen for prescription-only sale in 1976.71 The FDA did not remove the prescription requirement until 1994, 18 years later.72
- Compared to their British counterparts, U.S. consumers of naproxen were lucky. The United Kingdom did not approve naproxen for OTC sale until 2008, 14 years after the FDA did.73
To the extent the higher financial and time costs associated with obtaining prescriptions made accessing ibuprofen and naproxen prohibitive, government-imposed prescription requirements denied consumers access to these beneficial drugs.
Undermining Safety: The Case of Antihistamines
The case of antihistamines shows that government-imposed prescription requirements do not always promote safety and sometimes put patients at greater risk. In this case, the FDA used its power to mandate prescriptions in a way that steered patients away from safer drugs toward more dangerous drugs.
First-generation antihistamines such as diphenhydramine (i.e., Benadryl) and chlorpheniramine (i.e., Chlor-Trimeton) have been available OTC in the United States since the 1970s and 1980s.74 These drugs carry significant downsides. One USA Today article points out, “The recommended doses of these older antihistamines cause sedation that is the equivalent of being legally drunk.”75 In 2007, the Federal Aviation Administration reported, “Based on the information available in the [National Transportation Safety Board] Database [from 1990] through September 2006, the use of the antihistamine(s) by pilots was determined to be the probable cause or a contributing factor in 63 of the 338 accidents” that resulted in fatalities.76 Studies have also linked the drugs to thousands of automobile fatalities. In rare cases, they cause “seizures, hallucinations and death from accidental overdoses, especially in children.”77
Second-generation antihistamines are far safer. “Claritin [i.e., loratadine] causes no sedation and has no significant side effects. The drug is so safe that no lethal dose is known.”78 The FDA approved loratadine in 1993 and cetirizine and fexofenadine in 1996.79 Schering-Plough Corp., which manufactured Claritin, “used the drug’s safety record to persuade regulatory agencies in other countries to let the company sell the drug over the counter.”80
For years, however, the FDA steered patients toward the more dangerous drugs by letting them purchase first-generation antihistamines OTC but requiring prescriptions for the safer second-generation antihistamines. In 2001, an FDA advisory panel voted in favor of making loratadine, cetirizine, and fexofenadine available OTC,81 “but the FDA essentially disregarded the advice by failing to act.”82 Robert M. Miles, past president of the American College of Allergy, Asthma, and Immunology, noted, “The over-the-counter medicines are much more dangerous than the ones we write the prescriptions for.”84 In other words, the FDA got the safety considerations exactly backward.
The FDA dragged its heels for years, not switching loratadine (Claritin) to OTC until 2002, cetirizine (Zyrtec) until 2007, or fexofenadine (Allegra) until 2011.85 (For more about the circumstances of those switches, see the “Prescription Requirements Are a Boon to Pharmaceutical Manufacturers” section.)
A Decade of Denied Access to Emergency Contraceptives (“Plan B”)
Government-imposed prescription requirements politicize what should be personal medical decisions. One can see this effect most clearly in the decadelong struggle to respect the right of consumers to purchase levonorgestrel-based emergency contraceptives, commonly called “Plan B.”
Emergency contraceptives are now available OTC in the United States, including in vending machines and even via delivery services that some describe as “Uber for birth control.”86 But that was not always the case. For purely political reasons, Republicans and Democrats together denied consumers the freedom to self-medicate with Plan B for more than a decade. In total, the federal government took 12 years and 4 months—4,499 days—to make Plan B available without restriction. Even then, the switch came only after “a federal judge angrily accused the [Obama] administration of blocking the drug because of politics, not science, and ordered [Health and Human Services Secretary Kathleen] Sebelius to reverse her decision.”87
The FDA approved Plan B for prescription-only sale in 1999. In February 2001, the Center for Reproductive Rights and “more than 60 other family planning and health organizations” petitioned the agency to eliminate the prescription requirement.88 In 2003, an FDA advisory panel voted 23–4 to approve the petition.

In 2004, the FDA denied the petition. The nonpartisan U.S. Government Accountability Office (GAO) identified four ways the FDA’s denial was “unusual.”
First, the directors of the offices that reviewed the application, who would normally have been responsible for signing the Plan B action letter, disagreed with the decision and did not sign the not-approvable letter for Plan B. The Director of the Office of New Drugs also disagreed and did not sign the letter.
Second, FDA’s high-level management was more involved in the review of Plan B than in those of other OTC switch applications.
Third, there are conflicting accounts of whether the decision to not approve the application was made before the reviews were completed.
Fourth, the rationale for the Acting Director’s decision was novel and did not follow FDA’s traditional practices.89
The FDA’s assistant commissioner for women’s health, Susan Wood, resigned in protest of the unorthodox and unnecessary delays.90 Federal Judge Edward R. Korman would later summarize: “The 2003 FDA advisory committee formed to consider the first application for over-the-counter access to [Plan B] emergency contraceptives voted by the most overwhelming of margins to approve it.… It was only the political interference by the Bush White House that prevented their recommendation from being adopted.”91
In 2006, the Bush administration finally relented and allowed OTC sales for Plan B—but only to consumers age 18 and over. In 2009, a federal judge ordered the FDA to allow OTC sales to 17-year-olds.92
The struggle to give Plan B full OTC status seemed to reach a turning point in 2011, when FDA officials recommended unrestricted OTC access. Instead, Sebelius overruled the decision on the basis that there was insufficient data to show that 11- and 12-year-old girls would understand the Plan B label and use the drug properly.93 President Obama defended the decision, telling reporters that he shared concerns about selling Plan B “alongside bubble gum or batteries.”94
Critics—principally from the political left—accused Obama of violating a campaign promise and blocking unrestricted access to Plan B to aid his 2012 reelection campaign. They noted that the federal government had never applied such a standard to OTC drugs that had known harms, while Plan B by contrast is so safe that it is “impossible to overdose.”95 The editor in chief of the New England Journal of Medicine joined Susan Wood and another original member of the FDA committee that recommended removing the prescription requirement to write:
In our opinion, the secretary’s decision to retain behind-the-counter status for Plan B OneStep was based on politics rather than science. It cannot be based on issues of safety, since a 12-year-old can purchase a lethal dose of acetaminophen in any pharmacy for about $11, no questions asked. The only documented adverse effects of a $50 dose of levonorgestrel are nausea and delay of menses by several days. Any objective review makes it clear that Plan B is more dangerous to politicians than to adolescent girls. Thus, we once again have a situation in which political considerations are forming the basis of public health policy—resulting in another sad day for women.96
Left-leaning journalist Jonathan Cohn wrote, “It’s likely politics played a major role here. Most likely, the White House didn’t want critics—like, say, the eventual Republican nominee for president—saying that Obama wants 12-year-old girls to have sex. Obama wouldn’t be saying that, of course, but when has that ever mattered?”97
Despite intense public pressure, the delays continued until May 2013, when Judge Korman ordered the FDA to approve Plan B for unrestricted OTC sale. Korman wrote that the Obama administration blocked full OTC access for “obviously political” reasons:
The effort to convert these [Plan B] contraceptives from prescription to over-the-counter status has gone on for over twelve years, even though they would be among the safest drugs available to children and adults on any drugstore shelf. The FDA, responding to unjustified political interference, delayed as long as it possibly could before it took even one incremental step in the process. Ultimately, on December 7, 2011 … the FDA concluded that Plan B One-Step—the one-pill version of the drug—could be sold over-the-counter and without a prescription or age restriction. The FDA was reversed by the Secretary of Health and Human Services on the same day in a decision that was politically motivated and that, even without regard to the Secretary’s motives, was so unpersuasive as to call into question her good faith.98
Korman chided his own naiveté for thinking that the Obama administration’s “new FDA Commissioner, Deputy Commissioner, and President … could be ‘trusted to conduct a fair assessment of the scientific evidence.’”99 The agency complied with Korman’s order in June 2013.100
If government didn’t have the power to block or impose conditions on access to drugs, politicians would never have been able to politicize women’s health and reproductive rights in this manner. Manufacturers could just sell oral contraceptives directly to women and their partners. We will never know how many unwanted pregnancies and abortions occurred during those 12 years because the federal government denied those rights to manufacturers and consumers.
Wood warned that the FDA’s handling of OTC status for Plan B “could set a dangerous precedent for future decisions.”101 Indeed, it is not even an outlier in terms of the degree of political interference with the right of individuals to self-medicate. Politics continues to play a role in the FDA blocking OTC status for routine-use oral contraception and life-saving naloxone.
Routine-Use Oral Contraceptives
An ongoing example of government-imposed prescription requirements blocking access to beneficial medicines is routine-use oral contraceptives. Tens of millions of American women—more than four out of five women who have had sexual intercourse—have used oral contraceptives, which are critical for reducing unwanted pregnancies and the incidence of abortion.102 Obtaining a physician’s prescription can add up to $200 plus time and discomfort to the cost of “the pill.”103 Nearly a third of American women who seek prescriptions for oral contraceptives report having difficulty obtaining them, citing nonmonetary obstacles such as getting to their doctors’ appointments twice as often as they cite difficulty paying for the appointments.104
The Institute for Women’s Policy Research recommended switching the pill to OTC status as early as June 2000. “The public health benefits associated with reduced rates of unplanned pregnancies and abortions as well as the medical cost savings to society would substantially outweigh any risks of increased and undirected use of oral contraceptives,” the nonprofit organization wrote in 2001.105
Medical authorities have long endorsed OTC access to oral contraceptives. The American College of Obstetricians and Gynecologists (ACOG) has called for making the pill available OTC since at least 2012.106 In 2019, the ACOG issued an even more forceful call for OTC access and no age restrictions to “oral contraceptive pills, vaginal rings, the contraceptive patch, and depot medroxyprogesterone acetate” (i.e., contraceptive injections such as Depo-Provera).107 The American Academy of Family Physicians affirmed its support for OTC access in 2014 and again in 2019.108 A 2009 survey of nearly 500 reproductive health care providers found that 74 percent supported eliminating prescription requirements for oral contraceptives, contraceptive patches, and vaginal rings.109
Women around the world have OTC access to the pill. A 2013 study in the journal Contraception found that oral contraceptives are available without a prescription in more than 100 countries.110 Countries where women are free to purchase the pill without a prescription include communist China and Cuba. Countries that require prescriptions include the United States, Saudi Arabia, and most Western European countries.111
There is convincing evidence that women can use oral contraceptives safely and effectively without consulting a physician. Like all medications, oral contraceptives carry risks. Women who are smokers or have hypertension or other conditions are at a greater risk of adverse reactions. Yet oral contraceptives are unexceptional in this regard. Many widely accepted OTC medications such as aspirin, acetaminophen, and ibuprofen carry risks of adverse reactions.112
Indeed, eliminating the prescription requirement for the pill could lead to more careful management of those risks. A 2006 study of women in Seattle-area family planning clinics found that those who sought contraception took more careful account of potential contraindications than physicians do, suggesting OTC access would subject women to fewer risks.113 Research also suggests that prescription requirements contribute to contraceptive discontinuation within the first year and that allowing OTC access would make oral contraceptives more effective by promoting more continuous use.114
In 2020, however, neither the leading Democratic nor Republican bills that purport to provide OTC access to oral contraceptives would do so. The leading Republican bill would merely command the FDA to give the issue expedited consideration—without any consequences if the agency continues its current, dilatory approach. Worse, the bill would impose a statutory prescription requirement for minors, something that does not even exist for lethal doses of acetaminophen, aspirin, ibuprofen, and other OTC drugs.115
The leading Democratic bill is no better and may be worse. In service of their separate political goal of forcing insurers to pay for oral contraceptives, many Democrats and allied organizations oppose OTC status or have placed conditions on their support for it. Typically, they argue that OTC access could undermine the Affordable Care Act (ACA). The ACA requires nearly all Americans to purchase full coverage for all FDA-approved prescription contraceptives. If the FDA were to give oral contraceptives OTC status, insurers would no longer have to cover them. Critics have accused the organization Planned Parenthood of opposing OTC status because it could lose a significant share of its revenues if retailers could stock the pill on shelves alongside “bubble gum and batteries” and thereby provide women a lower-cost option.116
The leading Democratic bill, moreover, would not even ask the FDA to expedite consideration of OTC status. Instead, it would merely expand the ACA’s contraceptives-coverage mandate to require consumers to purchase coverage for all OTC methods of contraception, from hormonal contraceptives to condoms.117 It would do so even though such a mandate could block one of the main potential benefits of OTC status: lower prices for oral contraceptives. (See the “Excessive Coverage for Prescription Contraceptives Led to Price Spikes” section and especially Figure 4.) Rather than standing up for women’s rights, both Democrats and Republicans are playing politics with the pill, just as they did with Plan B. Congress (or the FDA) should switch oral contraceptives to OTC status immediately.
Blocking Access to Life-Saving Naloxone
Government-imposed prescription requirements prevent medical personnel and laypeople from saving lives. For example, the FDA requires consumers to obtain prescriptions before purchasing naloxone, a safe, effective drug that reverses opioid overdoses. The drug reverses depressed respiratory rate and blood pressure by knocking opioids off the recipient’s opioid receptors and binding itself to those receptors. The World Health Organization considers naloxone an “essential medicine.”118 It has no abuse potential.119 It has negligible or no effect on individuals who are not suffering an opioid overdose. It received a U.S. patent in 1961 and FDA approval in 1971.120

A World Health Organization article on the importance of making naloxone widely available.
The need for naloxone is dire. The U.S. Centers for Disease Control and Prevention (CDC) reports that from 1999 through 2017, nearly 400,000 U.S. residents died from opioid overdoses. Opioids account for more than two-thirds of total drug-overdose deaths, and mortality from opioid overdoses continues to rise.121
Naloxone is highly effective even when nonmedical personnel administer it. A 2013 study that trained opioid users and bystanders in Massachusetts found that “death rates from opioid overdose were reduced in communities where overdose education and naloxone distribution was implemented compared with not implemented.”122 The CDC reports that from 1996 through 2014, nonmedical personnel using naloxone reversed more than 26,000 opioid overdoses. The CDC says, “Providing opioid overdose training and naloxone kits to laypersons who might witness an opioid overdose can help reduce opioid overdose mortality.”123 In 2018, the U.S. surgeon general issued an advisory on the effectiveness of naloxone and urged its public distribution to combat the opioid-overdose crisis.124
There is no better way to put this safe, effective, life-saving drug within the reach of those who might witness an opioid overdose than to make it available to anyone who wishes to purchase it. Naloxone has been available OTC in Australia since 2016 and in Italy since 1996.125 Human Rights Watch writes, “Naloxone’s status as a prescription rather than an ‘over-the-counter’ medication creates a significant barrier to expanded access.” The organization continues, “It is therefore essential that prescription rules be changed … to designate naloxone as an ‘over-the-counter’ medication that can be issued without prescription.”126 According to one Human Rights Watch researcher, “If it were as easy to buy as Tylenol, many thousands of lives could be saved.”127
The FDA nevertheless continues to forbid the purchase of naloxone without a prescription.128 In September 2018, the FDA received a “citizen’s petition” from more than 70 health care practitioners and researchers asking the agency to approve easy-to-use naloxone nasal spray for OTC sale.129 As the FDA boasts of undertaking “unprecedented new efforts to support development of over-the-counter naloxone”—including taking the unusual step of drafting an acceptable OTC label for the manufacturer to submit back to the agency that “contains the information … a consumer needs to administer naloxone safely and effectively”—the agency writes that it “has been unable to reach a decision on [the] petition because it raises complex issues requiring extensive review and analysis by Agency officials.”130
Every day that the FDA continues to require prescriptions for naloxone, an estimated 130 U.S. residents die from opioid overdoses.131 Congress can and should eliminate the prescription requirement and make naloxone available for unrestricted sale.
Exacerbating Public Health Crises
Government-imposed prescription requirements jeopardize access to medicines during public health crises. During the COVID-19 pandemic, fear of disease and/or government-imposed public health measures (e.g., social distancing) have made complying with prescription requirements more difficult than usual. The British Pregnancy Advisory Service reported that women in the United Kingdom are “struggling to access contraception” because, “while [physicians] are supplying prescriptions, there is a long wait for telephone appointments which can mean that there is a gap during which they are not protected against unplanned pregnancy.”132 The ACOG writes, “COVID-19 response, including social distancing recommendations and delays to routine in-person visits, amplifies logistical obstacles to contraceptive initiation and continuation.”133
Even if government allows physicians’ offices and pharmacies to remain open, consumers may reasonably be afraid to patronize them. Some consumers may be able to obtain prescriptions electronically and/or purchase prescription drugs through online pharmacies. Yet those options may not work for people who lost their jobs or who live in jurisdictions that require in-person consultations before physicians can treat patients via telemedicine. Purchasing prescription drugs online can also create delivery lags and privacy concerns. Consumers stuck in unhappy housing situations may not want their contraceptives, naloxone, or other medications to arrive in the mail. OTC status for these drugs would eliminate delivery lags and privacy concerns by giving consumers the option to purchase them immediately at self-checkout registers and vending machines. An FDA-imposed prescription requirement further limits access to drugs during a pandemic by subjecting them to various dosage and time limits that states impose on prescription drugs.134
Prescription requirements can also become more problematic in unexpected ways. One Indiana police department announced that out of concerns of exposing its officers to SARS-CoV‑2, the virus that causes COVID-19, officers would no longer administer naloxone to overdose victims.135 If first responders refuse to administer naloxone for fear of contracting a disease, the prescription requirement becomes more burdensome and OTC access more urgent.
Many states’ temporary reforms during the COVID-19 pandemic illustrate how prescription requirements become more restrictive during a public health crisis. Several states suspended or eased licensing and scope-of-practice laws that restrict nurse practitioners from writing prescriptions, prohibit out-of-state physicians and nurse practitioners from writing prescriptions for state residents, and/or restrict prescribing via telemedicine.136 Such steps are an implicit admission that prescriptions become more difficult to obtain during public health crises. The ACOG recommends that states let physicians prescribe contraceptives via telemedicine without an initial in-person consultation, as some states require, and let pharmacists prescribe contraception for consumers of all ages.137 Such recommendations are necessary only because the FDA continues to require a physician’s prescription for most contraceptives.
Making certain drugs widely available without prescriptions, over the counter, at self-checkout registers, on the internet, and in vending machines could help mitigate public health crises. “If people have naloxone in their homes,” explains the New Jersey Harm Reduction Coalition’s Caitlin O’Neill, “they would be able to immediately have somebody reverse an overdose, as opposed to sort of having to be triaged for care during COVID-19, because we do need those first responders for the COVID patients.”138 Indeed, broad access to naloxone becomes more important when social-distancing measures lead to increases in isolation, depression, and opioid overdoses. In April 2020, Arkansas reported dramatic increases in naloxone reversals of opioid overdoses stemming from both recreational opioid use and suicide attempts.139 As the COVID-19 pandemic expanded the need for naloxone, prescription requirements constricted access.
In response to the pandemic, Congress did alter OTC drug regulation. Unfortunately, the steps Congress took were small, applied only to new versions of drugs that are already OTC, and often moved in the wrong direction. Congress reduced regulatory barriers for new versions of some OTC drugs but revoked the OTC status of other drugs, effectively removing them from the market. It required manufacturers of OTC drugs to pay “user fees” to the FDA and granted 18 months of market exclusivity to manufacturers of certain OTC drugs. Both provisions are likely to result in higher prices, further limiting consumers’ right to self-medicate. Congress took no steps to facilitate switching drugs from prescription-only to OTC status.140
A Contributor to Excessive Drug Prices
Evidence suggests that, in addition to the other access barriers they create, prescription requirements correlate with higher drug prices and that removing them correlates with reductions in drug prices.
One can see the first correlation in prices for ibuprofen and naproxen sodium. Per-milligram prices for these drugs are much higher above the dosage level where the FDA requires prescriptions. Given that consumers are free to substitute three nonprescription 200 milligram (mg) ibuprofen tablets for one prescription-only 600 mg tablet, or four 200 mg tablets for one prescription-only 800 mg tablet, one would not expect the per-milligram price of ibuprofen to vary much across these three types of tablet. Yet the per-milligram price for prescription-only ibuprofen is significantly higher than for OTC ibuprofen. Drugs.com indicates that the cash price for 40 prescription-only 600 mg ibuprofen tablets ($14.55) is 51 percent higher than the dosage-equivalent cash price for 120 nonprescription 200 mg tablets ($9.62). The cash price for 30 prescription-only 800 mg tablets ($12.86) is 34 percent higher.141
Prices for naproxen and naproxen sodium provide an even clearer illustration of a correlation between prescription requirements and higher prices. Per-milligram prices for OTC naproxen sodium are significantly lower than for prescription-only versions. Naproxen and naproxen sodium are close substitutes for each other, which implies that a dose-equivalent quantity of an OTC version of either drug would be a close substitute for the prescription-only version of either drug. The FDA requires prescriptions for 250 mg, 375 mg, and 500 mg naproxen tablets and for 275 mg and 550 mg naproxen sodium tablets but allows OTC sales of 220 mg naproxen sodium tablets.
Figure 1 shows that per-milligram prices for prescription-only naproxen strictly exhibit economies of scale. The per-milligram price for the drug falls both as the dose per pill increases (i.e., within each three-column cluster) and as pills per package increase (i.e., for each colored bar as one moves left to right along the X‑axis). Figure 2 shows that per-milligram prices for prescription-only versions of naproxen sodium similarly exhibit economies of scale. Yet per-milligram prices for naproxen sodium fall dramatically when the dose per pill is below the threshold where the FDA requires a prescription. The per-milligram price of prescription naproxen sodium is 3.3–4.2 times higher than for OTC naproxen sodium.142
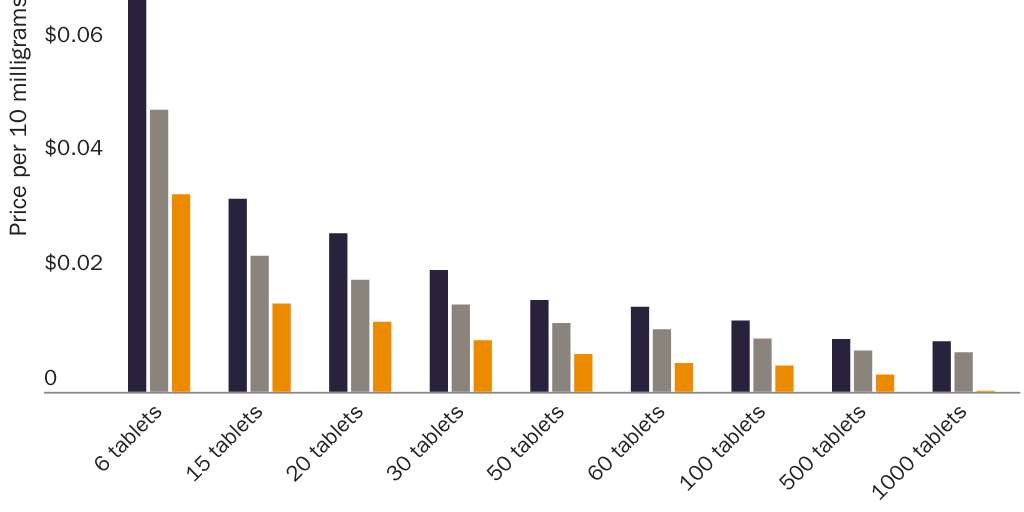
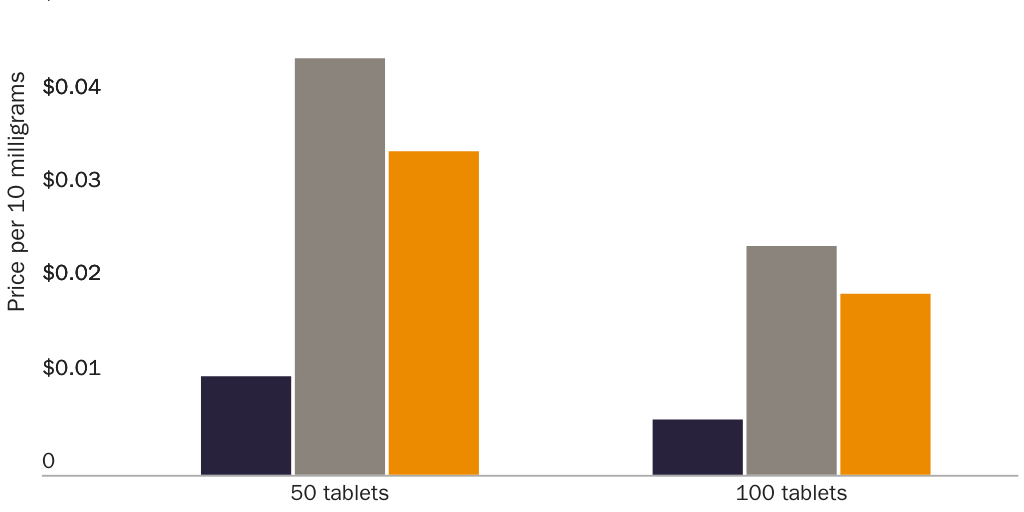
Numerous examples and studies show a correlation between removing prescription requirements and reductions in the price of a drug. In 2001, a month’s supply of Claritin (loratadine, 10 mg) was $11 in Canada, where it was available OTC, compared with $62 in the United States.143 After the FDA switched loratadine to OTC in 2002, the U.S. price fell below what many insured patients had been paying in copays.144 The U.S. price for a month’s supply is now about $7.50.145 In 2003, the FDA switched the proton pump inhibitor (PPI) omeprazole (Prilosec), an anti-heartburn medication, to OTC status. The price fell so much that when Arkansas’s state employee health plan started covering OTC omeprazole, the average price it paid across all PPIs—prescription and nonprescription—fell by 47 percent.146 A 2013 review of prescription-to-OTC switches that occurred between 1995 and 2010 found that the vast majority of studies (75 percent) “predicted cost savings for payers and patients.”147 In 2018, economist Sam Peltzman wrote, “The effect on prices is … clear: every study of the matter shows substantial price reductions when drugs move to OTC.”148
Do Prescription Requirements Increase Drug Prices?
It may seem that prescription requirements themselves cause drug prices to be higher and that removing them directly causes drug prices to fall. But the issue is more complex. Economic theory predicts that a prescription requirement will reduce a drug’s price and that removing it will increase a drug’s price.149 Removing a prescription requirement also subjects a drug to the higher taxes that Congress and state legislatures impose on OTC drugs; those explicit taxes somewhat mitigate the direct effect of removing the prescription requirement’s implicit tax.150 Patents and other forms of market exclusivity—whose purpose is to increase drug prices—apply to prescription drugs more often than to nonprescription drugs, and they often expire at the same time a drug switches from prescription-only to OTC. Claritin and Prilosec lost market exclusivity at approximately the same time the FDA allowed OTC versions.151
Third-party payment tends to increase drug prices. Insured consumers resist efforts by third-party payers to respond to high prices by switching to cheaper drugs or pharmacies, because the resulting savings appear to go not to the consumer but to an employer, an insurance company, or the government. “Insurers fear that, if they refuse to fork over the dough, their sick customers will be outraged.”152 Decisions by government and private insurers about whether and to what extent health insurance plans will cover a drug therefore have a major influence on drug prices. This inflationary effect occurs more often with prescription drugs than OTC drugs, because health insurance plans typically cover prescription drugs and stop covering them when an OTC version becomes available. As a result, “typically, prices for OTC products are lower … since consumers pay for OTC products directly, without any subsidization by insurance companies.”153
Various government policies, moreover, encourage excessive levels of drug coverage. Both tax policy (e.g., the tax exclusion for employer-sponsored health insurance) and government spending programs (e.g., Medicare and Medicaid) encourage excessive drug coverage by encouraging consumers to demand more drug coverage than they would if they were making level tradeoffs between drug coverage and other uses of money.154 Excessive levels of insurance can end up increasing prices for prescription drugs so much that the copayment that enrollees pay—which is only a portion of the insured price—can often end up higher than the total cash price. Consumer Reports writes:
If you have a standard insurance co-pay, it might not occur to you to shop around. But sometimes the price you’d pay out of pocket (what those without insurance are charged) might be less than your co-pay.… Metformin—used to treat type 2 diabetes—sells for just $4 for a month’s supply, or $10 for a three-month supply, at stores such as Target and Walmart, while a co-pay for a month’s worth averages about $11.155
Medicare also pays higher prices for drugs than enrollees could get by paying cash. One study found that for roughly 21 percent of Medicare Advantage and standalone Medicare Part D prescription drug plans, the prices that those insurers pay for drugs to treat cardiovascular disease are so high, the copayments alone are more than the $4 that Medicare enrollees would pay for those drugs if they purchased them with cash at Walmart.156 Excessive insurance can even increase the prices that cash-paying consumers pay. According to Consumer Reports, “Retailers intentionally set the list price very high so that there’s no chance it could undercut what they get paid by insurers.”157 Without excessive insurance, in other words, even cash prices could be lower.
Government further encourages excessive drug coverage by mandating that health insurance plans cover certain prescription drugs regardless of price. Congress requires insurers to cover all FDA-approved forms of prescription contraception with no enrollee cost-sharing, for example, and requires private insurers that issue standalone Medicare prescription drug benefit plans to cover all antidepressant, antipsychotic, anticonvulsant, transplant-related immunosuppressant, antiretroviral, and antineoplastic drugs—no matter how high manufacturers set the prices.158 Such mandates limit the ability of insurers to curb higher prices by reducing coverage (e.g., through higher enrollee cost-sharing) or removing drugs from their formularies.
Excessive Coverage for Prescription Contraceptives Led to Price Spikes
Prices for oral contraceptives provide an illustration. After Congress dramatically reduced the share of consumers who were conscious of the cost of oral contraceptives—and dramatically increased the share of consumers who are unconcerned with prices for those drugs and will therefore rebel against attempts by third-party payers to negotiate lower prices—prices for hormones and oral contraceptives skyrocketed.
In August 2012, the ACA began phasing in a requirement that nearly all private health insurance plans cover all FDA‐approved forms of hormonal contraception with no cost‐sharing. In addition, from 2014 through 2017, the ACA enrolled an estimated 5 million previously uninsured women of child‐bearing age in private insurance or Medicaid, which also covers oral contraception with no cost‐sharing.159 These changes transformed the market for oral contraceptives by making consumers almost completely insensitive to price increases. Figure 3 shows, for example, that the share of women with large‐employer coverage who faced zero cost-sharing when purchasing oral contraceptives rose from 4 percent in 2010 to 85 percent in 2014 and 90 percent in 2018.160 All by itself, ACA‐mandated coverage of contraceptives “account[ed] for nearly two‐thirds (63%) of the drop in out‐of‐pocket spending on retail drugs” across all consumers from 2012 through 2014.161
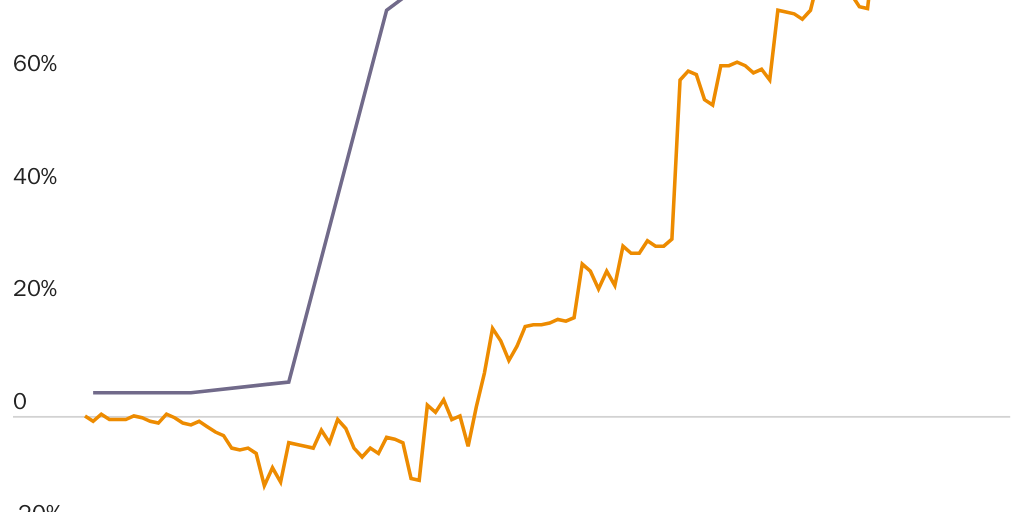
The ACA’s reshaping of the market for oral contraceptives coincided with a dramatic increase in prices for domestically produced hormones and oral contraceptives. Figures 3 and 4 show that from December 2009 through May 2013—a period prior to the ACA’s expansion of coverage for hormonal contraceptives and during which consumers more often paid for oral contraceptives directly—prices for hormones and oral contraceptives fell by 12 percent relative to inflation. Figure 4 shows price changes for these drugs generally followed a path like that of nonprescription drugs, which insurance typically does not cover and whose prices also fell in real terms.
When the ACA made oral contraceptives “free” for most purchasers, however, prices for hormones and oral contraceptives began to rise rapidly. By the time the mandate and coverage expansion took full effect in early 2014, prices for hormones and oral contraceptives had not only reversed those price reductions but matched the 17 percent growth in real prices for other prescription drugs. From May 2013 through May 2019, while real prices for nonprescription and prescription drugs overall rose just 12 percent and 37 percent, respectively, prices for hormones and oral contraceptives rose 108 percent—nearly three times the rate of growth for other prescription drugs.162
Removing Prescription Requirements Makes a Difference
The reason prescription requirements correlate with higher drug prices, and why removing such requirements correlates with price reductions, may therefore have more to do with excessive levels of health insurance than with prescription requirements per se. By itself, excessive drug coverage could explain the price differentials between dosage-equivalent quantities of prescription and nonprescription ibuprofen and naproxen sodium or the spike in prices for hormones and oral contraceptives that began in 2013. It could also help explain why Claritin’s price fell: not only did Claritin lose market exclusivity when the FDA switched it to OTC, but many insurers stopped covering it.163
Even so, although Congress and state legislatures have many options for expanding access to drugs and/or reducing drug prices, eliminating government-imposed prescription requirements is an important arrow in that quiver.164 Even if removing the requirements does not by itself directly reduce drug prices, it can help overcome other policies that increase prices—in particular, policies that encourage excessive insurance.
Removing the prescription requirement for just one drug in a class can increase price competition and reduce prices for substitutes for that drug. A survey of 12 managed care organizations found that when the FDA switched Claritin (loratadine) and Prilosec (omeprazole) to OTC, “All 12 organisations removed loratadine from their formularies and raised copayments for prescription antihistamines [e.g., Allegra and Zyrtec]. One third are taking all second generation antihistamines off their formulary. Eight removed omeprazole from the formulary, and seven raised the copayments for [all] prescription proton pump inhibitors.”165
Prescription Requirements Are a Boon to Pharmaceutical Manufacturers
A final and persuasive piece of evidence that prescription requirements enable drug companies to charge higher prices, and that eliminating them leads to reductions in drug prices, is the behavior of the pharmaceutical industry. Even though economic theory predicts that removing the implicit tax of a prescription requirement will increase sales, prices, and producer surplus, drug manufacturers actively resist efforts to have the FDA switch their products or their competitors’ products to OTC status. Drug manufacturers appear to know that, due to the interactions between prescription requirements and policies that encourage excessive levels of health insurance, prescription-only status lets them charge insurers higher prices and that OTC status will cause the prices of their products to fall.
Claritin and other second-generation antihistamines again provide a vivid illustration. In 1998, the health insurance company WellPoint petitioned the FDA to switch Claritin, Allegra, and Zyrtec from prescription-only to OTC status. WellPoint had special reason to request the switch: it expected to save about $100 million per year on claims for prescription antihistamines and related physician services.166
The drugs’ manufacturers all opposed the petition. As we previously noted, Claritin’s manufacturer Schering-Plough was aware that second-generation antihistamines are safer because it “used the drug’s safety record to persuade regulatory agencies in other countries to let the company sell the drug over the counter.” However, “in the United States, Schering-Plough oppose[d] selling Claritin over the counter, calling it unsound medical practice.”167
Critics alleged that the manufacturers’ opposition stemmed from another motivation: the desire to keep prices for their drugs as high as possible. A representative of a drug-industry trade group that represented Schering-Plough said, “Schering doesn’t want Claritin switched because it has wonderful profit margins now and doesn’t think it could come close to that in the OTC market.”168 An American University Law Review article claimed, “Schering-Plough, Aventis, and Pfizer are likely to generate millions of dollars in revenue if their drugs remain classified as prescription drugs. These companies admit that any loss of Rx status for Claritin, Allegra, and Zyrtec could seriously impact each company’s success.”169 The companies lobbied to preserve regulations that they knew were exposing consumers to harm—including deadly accidents—but that also boosted their profits.
In 2002, four years after WellPoint filed its petition, the FDA finally switched Claritin to OTC. The switch came not in response to WellPoint’s petition but because Schering-Plough reversed its position and itself petitioned the agency to make Claritin available OTC. The drug maker did this because Claritin’s patent was set to expire in late 2002, and the company had another second-generation antihistamine similar to Claritin that it was ready to promote in the prescription-only market. “By moving [Claritin] to the over-the-counter market,” the Washington Post reported, “the company hopes to dominate the prescription and nonprescription markets.”170
Such strategies still occur. As we previously discussed, naloxone is a life-saving drug with almost no side effects. In 2019, the Cato Institute announced that it would hold a briefing on Capitol Hill to educate Congress about the need to make naloxone available OTC.171 Shortly after Cato began publicizing the briefing, four different lobbyists representing the manufacturer of a nasal-inhaler version of the drug contacted four different Cato employees to press the manufacturer’s case for not switching this life-saving drug to OTC.
Even when manufacturers seek OTC status for their products, other manufacturers game the FDA’s powers for their own benefit. Anticipating the end of Prilosec’s market exclusivity, its manufacturer AstraZeneca petitioned the FDA to approve an OTC version, which would allow the company to use Prilosec’s brand recognition to gain a foothold in the OTC market. Manufacturers of generic versions of prescription omeprazole, including the company Andrx, “rightfully feared they would lose the opportunity to sell hundreds of millions of dollars of generic [prescription] omeprazole if most of the omeprazole market shifted to OTC.” In 2002, Andrx petitioned the FDA not to allow AstraZeneca to sell Prilosec OTC. The FDA denied the petition, but the fact that Andrx filed it delayed the agency’s approval of OTC Prilosec—and thereby forced Prilosec users to keep getting prescriptions—for seven months. Andrx benefited from the delay because “the longer generic omeprazole was the only way consumers could obtain low-cost omeprazole, Andrx … would continue to profit handsomely.”172
Corporate influence is yet another reason Congress should eliminate the FDA’s power to decide which drugs consumers need prescriptions to purchase. So long as government has this power, drug manufacturers will be major players in these decisions. Yet the interests of drug manufacturers conflict sharply with those of consumers. Manufacturers often have little incentive to submit or support OTC petitions and every reason to oppose them, because removing prescription requirements for their or their competitors’ products brings greater price competition and reduced profits. The FDA’s policy of waiting for manufacturers to initiate prescription-to-OTC switches therefore rigs the process against consumers. If the power to require prescriptions remains with the FDA, the agency should initiate switches itself and abandon its historical practice of waiting for the manufacturers to petition the agency to switch their products.
Americans Fare Poorly on Freedom to Self-Medicate
The U.S. system of drug regulation serves as a model for much of the developed world. Harvard professor Daniel Carpenter calls the system “perhaps the primary institutional export of the United States.”173 Even so, governments vary in the extent to which they require consumers to obtain prescriptions before accessing drugs, and U.S. consumers often fare poorly compared with consumers in other countries. In 2014, medicines-access consultant and pharmacist Natalie Gauld and colleagues wrote:
In contrast to her British counterpart, an American woman can now self-medicate for urinary incontinence. Conversely, without a prescription, this American woman cannot access a statin for her moderate cardiovascular risk, unlike in the UK; nor can she effectively treat her urinary tract infection, unless she is visiting New Zealand.174
Evidence suggests that the U.S. government may infringe on the right to self-medicate to a greater extent than governments in other nations. A 2009 GAO study shows that among the five nations and during the period studied, U.S. consumers were among those with the least freedom to self-medicate. The GAO examined government-imposed barriers to accessing 86 drugs in Australia, Italy, the Netherlands, the United Kingdom, and the United States. It found that foreign governments have created, in addition to OTC, at least four other categories of drug access that allow consumers to purchase those drugs without a physician’s prescription. The GAO’s descriptions of the categories appear verbatim in Table 1. Multiple countries had a “behind-the-counter” (BTC) category, which lets consumers purchase a drug with a pharmacist’s authorization. The “drugstore” category existed only in the Netherlands. The “OTC/pharmacist” category existed only in Italy.175

Figure 5 provides one perspective on the GAO’s findings. It groups the 86 drugs according to the level of permission a consumer needed from another person before purchasing the drug: “physician permission” (prescription), “pharmacist permission” (BTC), and “permission-less” (pharmacy, drugstore, OTC/pharmacist, and OTC). From this perspective, Australian consumers had the greatest freedom to self-medicate. Australians were free to purchase 52 (60 percent) of the 86 drugs without having to obtain permission from a government-appointed gatekeeper. U.S. consumers were free to purchase only 44 (51 percent) of the drugs without permission, a figure comparable to those for Italy (43; 50 percent) and the Netherlands (41; 48 percent). UK consumers were free to purchase only 29 (34 percent) of the drugs.
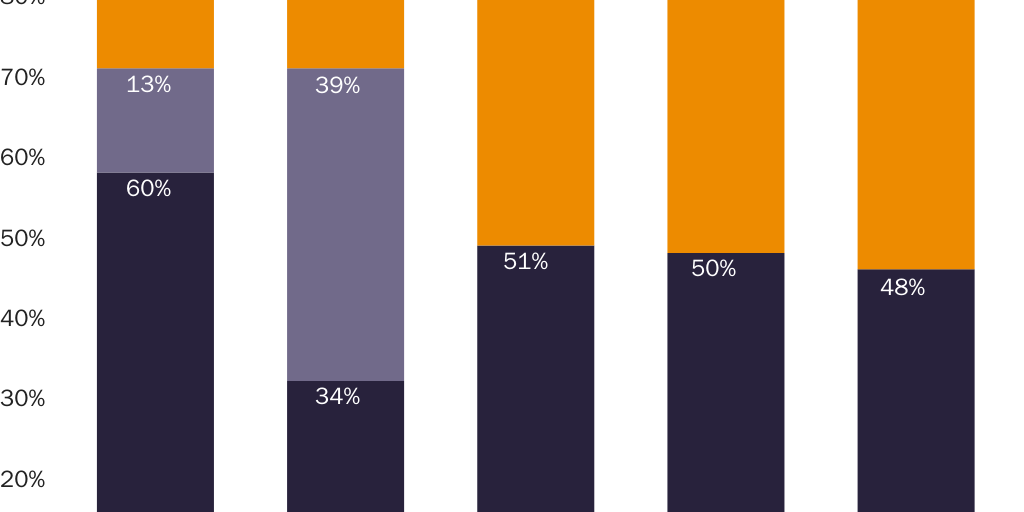
Whether UK consumers were overall more or less free to purchase these drugs than U.S. consumers depends on how one weighs requirements to obtain permission from a pharmacist. A “pharmacist permission” (i.e., BTC) requirement forces some consumers to have unwanted medical consultations. To the extent that those consultations discourage consumers from attempting to purchase the drug, lead to pharmacists denying permission to purchase the drug, or increase the time and monetary costs of purchasing the drug, BTC requirements could be akin to “physician permission” (i.e., prescription) requirements that leave consumers with less freedom to self-medicate. To the extent that those consultations are quick pro forma interactions or interactions that consumers would have had with one medical professional or another anyway, they could resemble permission-less access. If the United Kingdom’s BTC requirements were closer to the latter, UK residents had far greater freedom to purchase these 86 drugs than U.S. consumers. It means they could purchase 63 (73 percent) of these drugs with few or no government-imposed barriers, compared with 44 (51 percent) in the United States.
Regardless, Australian consumers were the freest. Even if Australia’s BTC requirements were as burdensome as prescription requirements, Australian consumers were still free to purchase 52 (61 percent) of the drugs without permission from a government-appointed gatekeeper. If Australia’s BTC requirements were merely pro forma, its consumers (like UK consumers) could purchase 63 (73 percent) of the drugs with few or no government-imposed barriers.
Another way to measure freedom to self-medicate is to consider the number of locations the government requires consumers to visit to purchase certain drugs. Figure 6 presents the GAO’s findings in terms of the number of “stops” a consumer needed to make to purchase each drug. “One-stop shopping” describes situations in which consumers had to make only one trip (i.e., to a vending machine, retail store, or pharmacy) to purchase a drug. “Two-stop shopping” indicates the government required consumers to make an additional and costly trip to a physician’s office. By this measure, consumers in the United States had considerably less freedom to self-medicate than consumers in Australia or the United Kingdom. Again, the degree of freedom in the United States was practically indistinguishable from that in Italy and the Netherlands.
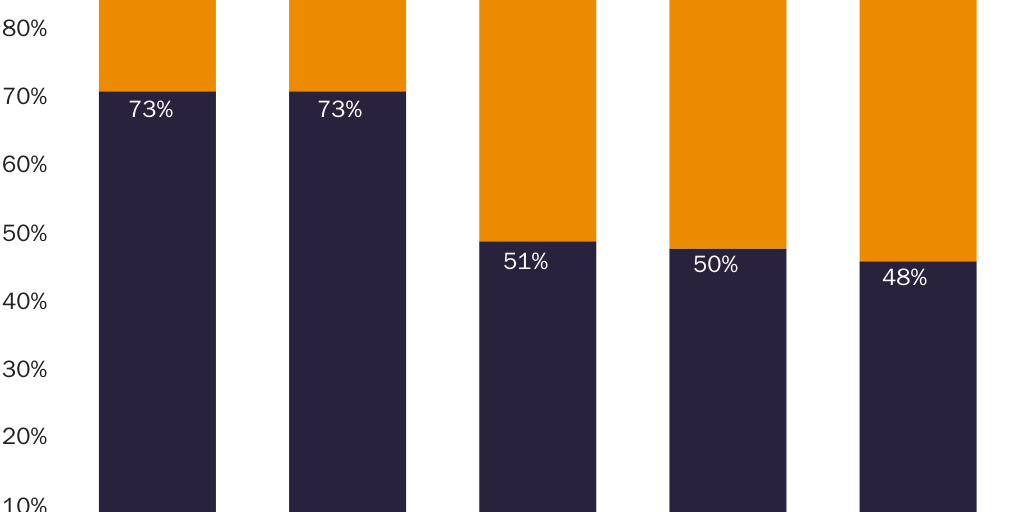
Oral Contraceptive Access across 147 Countries
Another indication that other countries impose fewer restrictions on the right to self-medicate than the United States is a 2013 study of 147 countries that found 70 percent allow consumers to purchase oral contraceptives without a physician’s prescription:
[Oral contraceptives] were informally available without prescription in 38 percent of countries, legally available without prescription (no screening by a health professional required) in 24 percent of countries, legally available without prescription (screening required) in 8 percent of countries and available only by prescription in 31 percent of countries … [Oral contraceptives] are available without prescription in the majority of countries.176
As this study notes, some countries may have prescription requirements but enforce them only weakly, with pharmacies ignoring the regulations and selling directly to self-medicating consumers. Researchers and journalists have documented poor compliance with prescription drug laws in many countries historically and currently in such countries as India, Hong Kong, Colombia, and Peru.177 Mexico only requires prescriptions for antibiotics and controlled narcotics. Many observers note a lack of compliance with prescription requirements for antibiotics.178
Falling Further Behind
Gauld and colleagues found that in recent years, the United States may have lost ground relative to other countries. They compared prescription-to-OTC switches in Australia, Japan, the Netherlands, New Zealand, the United States, and the United Kingdom from 2003 to 2013. The United Kingdom and New Zealand were the most aggressive in making “progressive” switches (i.e., removing prescription requirements). (Like the United Kingdom and Australia, New Zealand has a BTC category.) The United States, Australia, and the Netherlands were the least aggressive. The authors found, “Consumers in the more restrictive US and the Netherlands have continued to need to access doctors for a number of common medicines that have been switched in the UK and NZ.” For example, “a consumer in the United Kingdom could self-medicate with a non-sedating antihistamine 19 years earlier than a consumer in the United States.” Indeed:
Americans required a prescription for non-sedating antihistamines for considerably longer than all other countries, despite safety benefits over sedating antihistamines, which have long been non-prescription. Sedating antihistamines have been associated with workplace, car, and aviation accidents. Dutch women with vaginal candidiasis required a prescription for vaginal antifungals until 2011, 21 years later than in the [United States], with potentially unnecessary doctor workload, higher costs for the health funder, and prolonged discomfort and inconvenience for women.179
The authors concluded that consumers in the less aggressive countries “could be unnecessarily burdened by managing conditions that may reasonably be self-managed or pharmacist-managed instead.”180
Table 2 shows the current status of seven drugs across 21 countries. While not representative of all nations and less comprehensive than the GAO study, it shows substantial variation in prescription requirements. The vast majority—17—of the 21 countries allow consumers to purchase emergency contraception without a prescription. Four require a prescription. For daily-use oral contraception, the reverse is true: 18 countries require a prescription. Only three do not. In six countries, all in Europe, emergency contraception is the only one of the seven drugs available without a prescription. Hong Kong and Tanzania buck the trend by requiring prescriptions for emergency contraception but not for daily-use oral contraception. Belgium is the only country that requires prescriptions for both—indeed, Belgium is the only country that requires prescriptions for all seven drugs. Australia and Canada allow consumers to purchase four of the seven drugs without a prescription, more than the other countries. Yet, Australia and Canada each require prescriptions for daily-use oral contraception, while Australia requires prescriptions for insulin. Only four countries allow nonprescription sales of insulin, including countries such as the United States that only allow such sales for certain forms of insulin. Only four countries allow nonprescription sales of naloxone.

Finally, Table 2 shows that residents of other countries can self-medicate with many drugs that require a prescription in the United States. Consumers in the United States have less freedom to purchase these seven drugs than consumers in Australia and Canada. Salbutamol inhalers, which safely and effectively relieve asthma symptoms with few side effects, are available OTC in Australia.181 In the United States, they require a prescription.182 Consumers in a further nine of these countries (China, France, Hong Kong, Mexico, Singapore, South Africa, Sweden, Tanzania, and the United Kingdom) have more freedom than U.S. consumers to purchase at least one of these drugs. As we previously noted, consumers in communist China have more freedom to purchase daily-use oral contraceptives than consumers in the United States.
Do Government-Imposed Prescription Requirements Help or Hurt?
One might object that oral contraceptives, emergency contraceptives, and naloxone are cherry-picked examples and that a fair appraisal of government-imposed prescription requirements must also account for the benefits of saving consumers from more dangerous drugs. Before moving on to such an analysis, two counterarguments deserve attention.
First, even if government-imposed prescription requirements produce net health benefits—even if they improve health more than they harm health—they still violate the right of every person to determine what happens to her body. Just as we respect people’s right to refuse medical treatment even when they exercise it in ways that harm their health, we should respect their right to self-medicate even when they exercise it in ways that harm their health. Put differently, even if deceiving or coercing people into undergoing unwanted medical treatments would improve their health, it is wrong to deceive or coerce them into the treatments. Likewise, even if using coercion to prevent others from taking certain drugs would improve their health, it is wrong to keep them from obtaining the medications they want. The bioethicist Flanigan argues that if individuals have the right to take a drug intended to kill them—that is, to choose physician-assisted suicide—then they must also have the right to take any drug that poses far less risk to their health.183
Second, any burden of proof should lie not with those who support the freedom to self-medicate but with those who support using government coercion to stop people from taking the drugs they want. If anyone bears the responsibility of proving that their preferred rule passes a cost-benefit test, it is those who would put themselves in a position of controlling other people’s bodies and medical decisions. Curiously, supporters of government-imposed prescription requirements have never conducted such an analysis, neither before nor after depriving consumers of the freedom to self-medicate. It is disingenuous for either supporters of government-imposed prescription requirements or neutral observers to insist on a cost-benefit analysis before repealing such regulations but not before enacting them in the first place.
Government-Imposed Prescription Requirements Harm Health
Whatever one thinks of those counterarguments, the evidence suggests mandatory-prescription policies have the opposite of their intended effects: on balance, they harm health rather than promote it. Economist Peltzman performed several analyses to see if government-imposed prescription requirements correlate with improved health outcomes. In each analysis, such requirements had either no discernible effect or correlated with worse health outcomes.
First, Peltzman examined data on mortality from accidental or suicidal poisonings in the United States both before and after the FDA asserted the power to impose prescription requirements in 1938. He found that not only did government-imposed prescription requirements not correlate with reduced mortality from accidental or suicidal poisonings but that they instead correlated with increased poisonings.184 After controlling for other factors such as income, he found that “enforcement of prescription regulation increases poisoning mortality by 50 to 100 percent.”185
Second, he compared mortality from infectious disease in the United States before and after the introduction of prescription requirements for antibiotics. After again controlling for such factors as income and income inequality, he found “no remaining statistically significant difference in infectious disease mortality between countries that enforce prescription requirements for antibiotics and those that do not.”186
Finally, Peltzman compared the United States and other countries with government-imposed prescription requirements with countries that did not enforce prescription requirements. Again, the data showed that such requirements coincide with a higher incidence of poisoning:
Analysis of American time series suggests that regulation did not reduce—indeed, may have increased—poisoning mortality from drug consumption. An international comparison reinforces this suggestion: poisoning mortality is higher, all else remaining the same, in countries that enforce prescription regulation. This is consistent with other evidence that drug consumption in these countries is shifted toward more potent drugs.187
Peltzman’s analyses of drug poisonings only measured the effect of government-imposed prescription requirements on the harms caused by drugs. To the extent that such requirements also prevent patients from accessing beneficial drugs, the net effect is even worse than these analyses suggest.
How Could Consumer Safety Regulation Make Consumers Less Safe?
How could a regulation whose purpose is to protect consumers instead make them less safe? Peltzman offers two considerations. First, he notes that before the government began regulating pharmaceuticals, unregulated markets created mechanisms to protect consumers. Even before Congress imposed safety-testing requirements and the FDA began requiring prescriptions in 1938,
considerable progress had already been made in improving drug safety. The accidental poisoning rate had declined fairly steadily, even dramatically—by about two-thirds over the preceding four decades. This occurred in the face of gradually increasing drug consumption.… Thus the unregulated markets for drugs and for information about them had already substantially shrunk the problem that mandatory prescriptions were designed to deal with.188
In other words, Americans didn’t just sit around doing nothing while people died from drug poisonings.
Second, he postulates that prescription requirements increased poisonings because the presence of regulation caused consumers to be less careful than they were in the absence of regulation. At the margin, prescription requirements cause consumers to consult with physicians more often. Consumers’ confidence that someone else was looking out for their well-being made them less skeptical and more willing to try more dangerous drugs, which led to more adverse reactions. Peltzman writes, “The available data imply that, all else being the same, enforcement of prescription requirements raises per capita ethical drug consumption by over half. On average, ethical [i.e., prescription] drugs are more potent than over-the-counter drugs.”189 As a result, “there appears to be a moral hazard in this form of regulation much like that found in other forms of safety regulation: the regulation may lower the risk-cost per-pill, but this shifts consumption toward riskier pills.”190
To see how a greater reliance on physicians could lead patients to take greater risks, recall that a physician’s prescription is no guarantee of safety. Some 95 percent of the 105 Americans who died from elixir sulfanilamide and 100 percent of the 624 pregnant American women who took thalidomide took those drugs at the direction of government-licensed physicians. Investigators concluded that many doctors prescribed elixir sulfanilamide to patients who could not possibly have benefited:
Among the questionable conditions for which the drug was prescribed [were] “Bright’s disease, bichloride of mercury poisoning, renal colic, and backache,” none with the remotest connection to the infectious diseases for which sulfanilamide was known to work. Moreover, in “most cases” the recommended blood tests used to monitor patients on sulfanilamide “were not made.”191
U.S. physicians who prescribed thalidomide to pregnant women did so with little evidence of the drug’s effectiveness in combating morning sickness and “without telling them that the drug was an experimental one, making their patients the unwitting subjects of human drug experimentation.”192 In each case, many physicians did not even keep records of the prescriptions they wrote for these deadly drugs.193 Requiring consumers to consult with physicians is not always the safety-enhancing experience one might expect.
Economic historian Temin offers a third factor that helps explain how government-imposed prescription requirements could have the unintended consequence of increasing the risks consumers take and the harms they suffer: such requirements have made consumers more vulnerable to harm by making them more ignorant about health, medicines, and the nature of risk than they would have been without the government-imposed prescription requirements.194
When the FDA asserted the authority to require prescriptions for certain drugs, it not only removed those drugs from the consumer’s reach but actively denied consumers information about those drugs. In an effort to ensure that prescription-only drugs would only make their way to consumers through physicians, “the FDA would instruct firms to remove from their labels any remaining information that might guide lay users of prescription drugs.”195 Temin explains:
The regulation creating a separation between over-the-counter drugs (over which consumers have choice) and prescription drugs (about which consumers are supposed to know nothing) … deals with drug risks by attempting to deny consumers both the choice of dangerous drugs and the knowledge upon which such a choice might be based.…
[It] created a class of ignorant consumers as well as a class of prescription drugs. By specifying that information on prescription drugs be expressed in ways intelligible only to doctors, the regulation tried to prevent consumers from avoiding the regulation. But … consumers cannot be excluded from the decision to buy drugs, much less from the decision to take drugs. The result of the regulation therefore is to make [the] consumer’s participation in these decisions ill-informed and, and least legally, irresponsible. Phrased differently, some part of the gap between the drug knowledge of the average doctor and the average consumer is the product of regulation. This needs to be kept in mind when the magnitude of the gap is exhibited as an argument for more regulation.196
As public health professor Julie Donohue notes, “A paradoxical situation developed in which potentially dangerous prescription drugs were dispensed to consumers with less accompanying information than OTC drugs carried.”197
Had the FDA never claimed the power to create a class of prescription drugs, and had Congress never codified those powers, manufacturers would have sold more drugs directly to consumers and would have invested greater resources in educating consumers about the costs and benefits of their and their competitors’ drugs. The product of those marketing expenditures would be that the typical consumer would have become more knowledgeable about health, medicines, and the nature of risk.198
In sum, government-imposed prescription requirements make consumers more ignorant and less safe by replacing a more effective form of consumer protection with a less effective form, in part because government often prioritizes values other than consumer safety.
Governments Require Prescriptions for Reasons Other Than Safety
Gauld and coauthors note one final factor relevant to whether, on balance, government-imposed prescription requirements leave consumers more or less safe: safety concerns cannot explain why some governments take so long to give consumers direct access to many medicines. They write,
Considering the variation in switching medicines with a benign safety profile and limited risk of masking serious conditions, e.g. non-sedating antihistamines and mast cell stabilizers, our data suggest that factors that are not safety-related may delay switch.199
When governments wield the power to impose prescription requirements, they routinely base those decisions on factors other than safety.
The sole rationale for government-imposed prescription requirements is to increase consumer safety. Yet examples of government using this power to prioritize factors other than safety abound. They include the FDA blocking access to Plan B emergency contraception for 12 years; the agency’s continued opposition to OTC status for daily-use oral contraception; an entire decade when the FDA pushed consumers toward hazardous sedating antihistamines by requiring prescriptions for safer, nonsedating antihistamines; and the agency’s continued refusal to switch naloxone to OTC. When governments impose or preserve prescription requirements for reasons other than safety, they harm patients by blocking access to drugs that patients could otherwise afford and use safely.
The fact that governments base decisions about prescription requirements on factors other than safety undercuts the entire rationale for allowing government to wield this power in the first place. It is comforting to imagine that the power to impose prescription requirements on manufacturers and consumers will only fall into the hands of wise policymakers whose sole consideration is a selfless concern for consumers. The reality is not so idyllic. To the extent that government-imposed prescription requirements block access to beneficial medications, they leave consumers less safe, not more.
Eliminate Government-Imposed Prescription Requirements
In the interest of expanding access to pharmaceuticals and restoring the right to self-medicate, Congress should strip the FDA of any power to impose prescription requirements. This would require repealing the Durham-Humphrey Amendment and prohibiting the FDA from using other provisions of the Food, Drug, and Cosmetics Act of 1938 (FDCA) to impose prescription requirements, as the agency did prior to 1951. State legislatures could restrict medication access by minors and the cognitively disabled and, if necessary, enact specific limitations on access to certain antibiotics.200 If wholesale repeal of government-imposed prescription requirements is not politically feasible at present, Congress and state legislatures could take several intermediate steps that would reduce interference with the right to self-medicate and that could illustrate the benefits and wisdom of full protection of that right.
Prescriptions and Physician Consultations Would Continue
What would change if Congress were to eliminate the FDA’s power to impose prescription requirements? The available evidence suggests that any change would not be as much as one might think and that, on balance, it would be for the better.
One way things would not change is that consumers would continue to solicit the advice of physicians before consuming drugs. Flanigan predicts:
Imagine a world where people took rights of self-medication as seriously as they currently take informed consent requirements. Just as most patients continued to take their doctor’s advice after the adoption of informed consent requirements, most patients might continue to consult medical experts before using unauthorized and untested drugs, even if they were legally permitted to decide differently.… Rights of self-medication would change medical practice only in those cases where a patient’s judgment departs from a medical expert’s.201
Another way things would not change is that manufacturers would continue to sell many drugs on a prescription-only basis. The complexity, sophistication, and risks posed by many modern pharmaceuticals, and the threat of tort liability, would drive manufacturers to continue to market many drugs as prescription-only.202 The same liability fears that made manufacturers not want to label drugs for laypeople’s use when the federal government required it in 1938 would also guide their decisions today. Temin explains why prescription requirements would likely survive a repeal of government regulations requiring them:
Drug companies … are wary when the threat of litigation is present. Their experiences with suits arising from adverse side reactions do not make them anxious to increase their exposure by selling powerful drugs on the over-the-counter market.… Drug companies are exceedingly sensitive to the costs of being sued for the apparently negligent marketing of their products. In addition to the legal costs of defending themselves, the damage to their reputation may affect the sales of other drugs.203
Drugs for which manufacturers might continue to require prescriptions include antibiotics, which can affect third parties, and dangerous drugs such as phenobarbital, thalidomide, and narcotics.
The available evidence supports Flanigan’s and Temin’s predictions. Temin writes, “It had been possible to utilize prescriptions before 1938, but it also had been possible to buy virtually any drug without a prescription.”204 Yet consumers routinely sought the guidance of physicians, and prescriptions were ubiquitous, even when there were no regulations requiring either. Again, Peltzman notes, “About one-third of drug purchases were being made under a doctor’s prescription at the time the 1938 act was passed.”205 And again, 95 percent of Americans who died from taking elixir sulfanilamide did so under the supervision of a physician, even though no law required them to do so.
Broader Access
The principal way things would change if the FDA no longer had the power to impose prescription requirements is that access to many drugs—such as insulin, oral contraceptives, and naloxone—would expand. Broader access would result not only from manufacturers selling directly to consumers but also from lower prices and more convenient avenues for accessing those drugs.
Removing unnecessary prescription requirements would relieve consumers of the unnecessary time and financial costs of visiting a doctor to obtain a prescription and waiting for a pharmacist to fill it.206 Though consumers can obtain prescription drugs only through pharmacies, they have many convenient options for purchasing OTC medications, including online services, convenience stores, newsstands in airports, grocery stores, vending machines, etc.
One cannot predict exactly which prescription drugs would shift to OTC. It is possible or even likely, however, that elementary and secondary schools could more easily stock asthma medications and epinephrine injectors. Diabetics could purchase insulin in bulk via online retailers such as Amazon. Naloxone could become more widely available at vending machines and in first-aid kits. Places opioid users frequent might make naloxone available for emergency use, just as many establishments make defibrillators available.
Eliminating the unnecessary barriers would expand access the most for low-income consumers who struggle to pay medical bills and who might otherwise forgo treatment.207
Innovation
Today, innovators devote energy to reducing the cost of complying with unnecessary regulatory barriers to drug access. The Kaiser Family Foundation reports that one example of such innovation is services that help consumers overcome the regulatory barriers to purchasing oral contraceptives:
A growing number of online services and smartphone applications offer options for patients to speak with providers by video or chat, get prescriptions, and order birth control pills through mail delivery. These services work by collaborating with physicians, pharmacies, and sometimes health insurers to prescribe and ship [oral contraceptive pills] to the patient’s home or a local pharmacy.208
Eliminating government-imposed prescription requirements would not only expand access to needed drugs but would also free manufacturers, pharmacies, consumer advocates, and products-liability insurers to focus instead on innovations that educate consumers about health, risk, and how to use drugs safely and effectively.
Manufacturers, pharmacies, and insurers could develop new rules or drug-marketing categories that, for certain drugs, might strike a better balance between access and safety than Durham-Humphrey’s binary prescription/OTC structure. These marketing categories could include the BTC, pharmacy, drugstore, or OTC/pharmacist categories described in Table 1 as well as other potential categories. The new classifications could tap the often-underused knowledge and skills of, and provide new employment opportunities for, pharmacists and other clinicians.209 Manufacturers could require prescriptions but specify that clinicians other than physicians—including pharmacists, nurse practitioners, physician assistants, registered nurses, and psychologists—may provide the necessary authorization.
Manufacturers or pharmacies could establish procedures in which they agree to sell certain drugs directly to consumers but only if the consumers read or listen to safety information and/or demonstrate that they understand the information. Innovators have already developed pharmaceutical kiosks that remotely connect consumers to a live pharmacist.210 The businesses could create automated touchscreen kiosks that help consumers decide whether a drug is right for them. The kiosks could show interactive educational videos and instruct and test consumers on the safe use of drugs. They could record consumers’ answers to such questions, as well as information about the consumers’ health, including real-time blood pressure, pulse, and weight readings. The kiosks could photograph consumers if manufacturers or pharmacies consider it necessary to track who is purchasing certain drugs. They could provide comprehensive written instructions for patients to take with them. Kiosks could do all these things in multiple languages.
One cannot predict what other innovations drug manufacturers and pharmacies may develop, nor which innovations will prove cost-effective. These possibilities nevertheless illustrate how innovators might broaden access by developing consumer safety tools that do not exist today.
Congress should consider eliminating the FDA’s authority to mandate prescriptions as a way of preparing for future public health crises. Had government never acquired the power to impose prescription requirements, we could by now be enjoying the fruits of decades’ worth of innovation and consumer education. One cannot know for sure, but pharmaceutical vending machines and interactive kiosks could have been far more established and widespread at the beginning of the COVID-19 pandemic and could therefore have enabled consumers to access the drugs they need while minimizing the risk of transmitting disease.
Letting manufacturers decide whether to sell drugs directly to consumers or to require a prescription would inevitably lead to situations where for the same types of drugs, some manufacturers would choose the former and others would choose the latter. As it did in the past, this could cause confusion for some consumers and pharmacists.
Fortunately, there are several readily available solutions. In the simplest case, pharmacies could require a prescription for all versions of a given drug if even one manufacturer does so. Alternatively, pharmacies or their liability insurers could simplify matters for their customers by developing their own prescription requirements based on their assessments (and liability concerns) about whether a drug is safe enough for consumers to use without physician supervision. In addition, innovators could develop new decision aids. Manufacturers, pharmacists, and consumer advocacy organizations such as Consumer Reports all have incentives to develop tools to help consumers and pharmacists navigate different manufacturer rules. Finally, to the extent that consumers are as risk-averse as Peltzman and Temin argue, any confusion about whether they need a prescription would itself lead consumers to seek a physician’s advice, even in cases where there is no legal requirement to do so.
Consumers today have far more drug information at their fingertips than they did before the federal government created the current system of mandatory prescriptions. Modern information technologies can address divergent manufacturer rules without blocking access to beneficial medicines.
Savings for Consumers and Taxpayers
Eliminating prescription requirements can deliver additional benefits by reducing prices, eliminating unnecessary medical expenditures, and boosting economic productivity.
Removing prescription requirements can trigger greater price competition by exposing drug manufacturers to a broader population of more cost-conscious purchasers. Peltzman writes, “The effect on prices is … clear: every study of the matter shows substantial price reductions when drugs move to OTC.” He continues, “Of course, total cost—including the cost of physician visits and the value of the time and trouble of securing prescriptions—declines even further when the drug moves to OTC.”211
Scholars at the Brookings Institution have catalogued several ways that removing government-imposed prescription requirements could reduce health expenditures:
Cost savings might result from reducing the number of unnecessary physician visits or medical encounters solely used to obtain prescriptions. Emergency room visits may also be reduced as a result of increased access to early and appropriate treatment for acute symptoms. Following the switch of certain prescription medications, a significant decrease has been observed in the number of physician visits, laboratory charges, and prescriptions dispensed for enrollees. This has the potential to decrease medical expenditures for patients, payers, and managed care organizations.212
A 1992 review of the economics literature found that “the benefits, to individual consumers and to society as a whole, resulting from a change in prescription status outweigh the costs. The extent of the benefits depends on the type of drug and the size of the market.”213 Temin estimates that the FDA’s efforts to switch cold medications to OTC between 1976 and 1989 reduced the number of doctor’s visits by 1.6 million, saved U.S. consumers more than $142 million, and delivered net benefits of $1.6 billion in 1989 alone.214
With regard to productivity, one study estimated that if France, Germany, Italy, Poland, Spain, and the United Kingdom were to switch triptans—a class of medications that treats migraines—from prescription-only to OTC status, they would reduce spending on migraine care across those countries by 13 percent. The overall benefits would be even greater because better treatment of migraines would increase worker productivity. The estimated productivity gains accounted (coincidentally) for 13 percent of total estimated annual benefits of $188 million.215
Insurance Coverage of OTC Drugs
Critics object that moving drugs from prescription-only to OTC status might inhibit access because insurance generally covers prescription drugs but not OTC drugs. They say that if consumers face a drug’s full price without assistance from an insurance company or government, eliminating the prescription requirement may reduce access. While valid, such concerns do not justify retaining government-imposed prescription requirements. There are benefits as well as costs when insurers stop covering certain drugs, and insurers can (and often do) cover OTC drugs when doing so is cost-effective.
First, as we previously discussed, insurance coverage often increases drug prices, and removing prescription requirements can cause drug prices to fall so far that consumers can end up paying less than they had been paying in copayments. Antihistamines and diabetes drugs covered by private insurers and cardiovascular drugs covered by Medicare plans often have cash prices that are lower than the copays that insurance companies require enrollees to pay.216
Second, removing prescription requirements and thereby eliminating insurance coverage could in many cases reduce wasteful and inefficient drug consumption. Insurance coverage has a moral-hazard effect. It encourages enrollees to consume medical care whose value does not justify its cost. To the extent that prescription-to-OTC switches force consumers to pay more out of pocket for drugs, the additional costs could lead consumers to make more careful drug-consumption decisions and help to mitigate the growing problem of patients consuming unnecessary drugs.217 Since all drugs carry risks, this effect could also improve health by reducing the harms those consumers suffer, often for little benefit.
Third, insurers can cover nonprescription drugs if that’s what their customers want. The Brookings Institution reports:
Numerous state and private insurance providers have offered nonprescription drug coverage for medications that have switched from Rx-to-OTC. For example, an Arkansas state employee health plan has chosen to include coverage of nonprescription omeprazole (e.g., Prilosec), a medication used to treat heartburn, ulcers, and acid reflux disease. Research has demonstrated that coverage for this OTC medication produced cost savings and resulted in small increases in utilization.218
Employers have also implemented coverage for OTC allergy medications.219 Employers have incentives to cover OTC medications when doing so would reduce absenteeism and presenteeism (working while sick). Insurers have incentives to offer coverage of cost-effective OTC medications, because doing so can save insurers money and give them a quality-based competitive advantage.
Safer Consumers, Better Health
In the end, many people will judge proposals to eliminate government-imposed prescription requirements on how repeal would affect consumer health and safety. Fortunately for the right to self-medicate, the evidence suggests that eliminating these requirements on balance would result in greater safety and better health for consumers.
Principally, to the extent that repealing federal prescription requirements would cause consumers who would have consulted a physician to instead self-medicate, those consumers would generally consume less-risky drugs than they would under the guidance of physicians. Peltzman writes:
Let us, for a moment, think the unthinkable and suppose that we did away entirely with the regulation. What would happen? Most likely … the number of deaths from drug abuse would decrease. This is not because consumers are receiving inadequate advice from their doctors. Instead, it is because of the apparent conservatism of many consumers: if given the option of not seeking a doctor’s advice first, the evidence suggests that consumers are likely to shift from consuming more potent ethical drugs toward consuming generally less potent over-the-counter remedies.220
Once again, a physician’s prescription is no guarantee of safety.
At the same time, reducing the barriers to accessing beneficial medicines would increase the number of patients who could obtain treatment. Temin found that switching cold medicines and topical corticosteroids from prescription-only to OTC expanded access to such drugs and improved outcomes for people with colds.221 The study of triptans across six EU countries estimated that 46 percent of OTC purchasers would be “patients that are currently treated with partially effective OTC analgesics or those who are not treated at all.”222 In his study of cough and cold remedies, Temin suggested there are even “gains to be realized from switching more powerful drugs from prescription to OTC.”223
Removing government-imposed prescription requirements would also improve health by providing consumers relief faster than the existing system can. If consumers suspect they are having a migraine, they may have to wait days before they can secure a doctor’s appointment and fill a prescription. If they were able to purchase triptans OTC, it could spare them days of agonizing pain and lost productivity.224
Just as government-imposed prescription requirements have reduced consumers’ health literacy, removing them would improve their health literacy. The more manufacturers sell drugs directly to consumers, the more they will invest in educating consumers about their products and how to use them safely. The study of triptans in six EU countries notes that beyond the immediate health improvements from broader access as a result of OTC status, “it is possible to further reduce the disease burden, if manufacturers invest in a disease awareness campaign that will promote awareness of the condition to currently undiagnosed patients (40% of all migraine sufferers).”225
There is no way to structure access to drugs that would ensure that every individual receives every drug whose benefits exceed its costs and that no individual receives a drug where the reverse is true. Perfection is not an option. Eliminating government-imposed prescription requirements would lead to situations in which consumers misdiagnose their own ailments and take a drug that does them more harm than good. For example, triptans carry a small risk of cardiovascular events.226 The FDA places a strong warning on the labels for smaller doses of (prescription) sumatriptans than the United Kingdom allows consumers to purchase without a prescription.227 The study of six EU countries estimates that the savings and productivity gains from OTC triptans would exceed the additional expenditures and productivity losses from added cardiovascular events by more than 100-fold, but it does not attempt to estimate the health losses that would occur other than describing them as “rare.”228
Whatever the added risk, the way to minimize that risk and those losses is not to strip consumers of their autonomy, increase the price of their medications, and force migraine sufferers to endure unnecessary pain. It is to educate consumers about safe drug use and the associated risks so that they can make informed decisions. Consumers’ inherent risk aversion plus innovations in consumer education would improve consumers’ health literacy and reduce adverse drug reactions while broadening access to beneficial drugs. In cases where consumer education does not reduce those risks to acceptable levels, tort liability would force manufacturers to require prescriptions.
Reconsider Premarket Approval Requirements
Merely repealing Durham-Humphrey would not eliminate the FDA’s ability to impose prescription requirements. It could even backfire by leaving consumers with less freedom to self-medicate, because the FDA could still use other tools at its disposal to maintain its current paternalistic approach to pharmaceutical access. As it did in 1938, the FDA could use the FDCA’s labeling requirements to create de facto prescription requirements. It could claim that to approve certain drugs as safe to market to consumers that it must require more and larger safety and efficacy studies. It could extract explicit or implicit commitments from manufacturers to sell drugs as prescription-only in exchange for quicker approvals. If Congress did nothing more than repeal Durham-Humphrey, the FDA could exacerbate the problems of drug lag and drug loss and thereby reduce rather than expand U.S. residents’ freedom to self-medicate. Congress must therefore make additional changes to the FDCA, including amending the statute’s labeling requirements, to ensure that the FDA does not once again defeat congressional intent by claiming the power to require prescriptions.
Even with such changes, the FDA would retain the power to deny consumers their right to self-medicate through its control over the length and cost of its approval processes. That power stems from the FDCA’s requirements that all new drugs, all generic drugs, and all forms of manufacturer speech about new indications for existing drugs must receive the FDA’s approval before going to market. So long as Congress allows the FDA to act as a paternalistic gatekeeper between consumers and drugs, the agency will err on the side of delaying and denying consumers access to new products and new information.229
The case for eliminating government premarket approval requirements, like that for eliminating government-imposed prescription requirements, is strong. In the absence of FDA certification that drugs are safe and effective, private-sector organizations would perform that same function. The crucial difference is that private certification organizations would provide consumers information that they need to make educated treatment decisions rather than deny them access to medical care.
Drug Testing and Certification That Respects Health Care Rights
Private testing and certification of safety and efficacy predate the FDA and have always existed alongside it. Organizations such as the U.S. Pharmacopeial Convention, the American Medical Association (AMA), Consumer Reports, medical journals, health insurance plans, foreign regulatory bodies, and others have offered or continue to offer alternative safety and efficacy certification. The FDA itself reports:
In 1905, the AMA formed its own Council on Pharmacy and Chemistry which levied a fee on manufacturers to evaluate their drugs for quality (ingredient testing) and safety. Drugs accepted by the Council could carry the AMA’s Seal of Acceptance and only products with the seal had access to the advertising pages of the Journal of the American Medical Association (JAMA). The AMA’s Chemical Laboratory tested commercial statements about the composition and purity of drugs in their labs, while the Council on Pharmacy and Chemistry followed up with safety evaluations and rudimentary efficacy evaluations designed to eliminate exaggerated or misleading therapeutic claims.… The AMA’s drug certification program remained in place until 1955.230

The American Medical Association’s Council on Pharmacy and Chemistry evaluated data submitted by pharmaceutical manufacturers on the drugs they produce. Source: National Library of Medicine.
The Pharmaceutical Market Where All Certification Comes from outside the FDA
There is a consensus in American politics that the FDA should not be the sole authority on which drugs are effective for which uses. In the market for off-label uses for drugs, all efficacy certification comes from organizations other than the FDA. The FDA approves drugs to treat specific conditions and permits manufacturers to print only those approved indications on a drug’s label. Once a drug reaches the market, however, consumers and practitioners are free to use it in ways that the FDA has not approved, including for other conditions. Using a drug for a non-FDA–approved indication is an off-label use because the FDA has not (or at least not yet) allowed that use to appear on the drug’s label. Just as physicians are free to prescribe prescription-only drugs for off-label uses, consumers are free to use OTC drugs for off-label uses.
Off-label drug uses are common. An estimated 21 percent of prescriptions in the United States are off-label, with the share rising as high as 83 percent for individual drugs.233 They can be beneficial. The FDA approved thalidomide to treat leprosy and multiple myeloma only after doctors had started prescribing it off-label to treat those illnesses.234 The FDA itself acknowledges, “Aspirin has been shown to lower the risk of heart attack and stroke in patients who have cardiovascular disease or who have already had a heart attack or stroke … even though the directions on the aspirin label do not apply to this use of aspirin.”236 Some of the deaths from elixir sulfanilamide likely stemmed from some doctors prescribing it for off-label uses.237 (See the “How Could Consumer Safety Regulation Make Consumers Less Safe?” section).
Off-label use is controversial, in part because the amount of evidence supporting off-label uses varies. A 2006 investigation estimated, “Among off-label [prescriptions], most (73 percent) lacked evidence of clinical efficacy, and less than one third (27 percent) were supported by strong scientific evidence.”238 One study found that compared to patients who used drugs for FDA-approved (“on-label”) indications, those who used drugs for off-label indications are 44 percent more likely to suffer an adverse drug reaction and 54 percent more likely if the off-label use lacked strong scientific support. If an off-label use had strong scientific support, however, patients were no more likely to suffer adverse drug reactions than those who used drugs for on-label indications.239
With regard to evidence, patients, doctors, insurers, and even governments routinely rely on sources other than the FDA to certify the efficacy of off-label uses. Examples of non-FDA efficacy certification include drug compendia, Consumer Reports, foreign regulatory agencies, and medical journals.
In particular, the federal government and all 56 state and territorial governments recognize multiple voluntary, private-sector efficacy certifications as reasonable alternatives to the FDA’s seal of approval. Medicare, Medicaid, and the Children’s Health Insurance Program are the largest purchasers of pharmaceuticals in the world, covering more than 121 million individuals, or more than one-third of the U.S. population.240 Federal law requires the Medicare program to rely on specific, privately compiled drug compendia to certify the efficacy of off-label uses. It further authorizes the secretary of the Department of Health and Human Services (HHS) to designate additional compendia and medical journals on which Medicare must rely. Over time, HHS secretaries have accepted four compendia and more than a dozen medical journals as authorities on the efficacy of off-label uses.241 Federal law imposes similar requirements on state Medicaid programs.242
According to the American Society of Clinical Oncology, “Following the lead of the Congress, the overwhelming majority of state legislatures have enacted statutes requiring [privately insured consumers to purchase] coverage of off-label uses of cancer drugs based on the compendia.”243 As of 2009, 74 percent of the U.S. population resided in states that required consumers to purchase coverage that relies on similar sources of off-label efficacy certification.244
The FDA itself used a private, third-party reviewer—the National Research Council of the National Academy of Sciences—to evaluate and certify the efficacy of drugs the agency had approved based on safety alone between 1938 and 1962.245
Alternative sources of formal and informal off-label efficacy certification also move faster than the FDA. In a study of off-label indications for existing drugs, former U.S. Bureau of Consumer Protection director Howard J. Beales found:
In the average market, a majority of the [medical] journal articles discussing the new use had already appeared two years before [FDA] approval. At the time of approval itself, two-thirds of the journal articles had already appeared. Moreover, the new uses were recognized in U.S. Pharmacopeia Drug Information, an authoritative compendium of prescription drug information, an average of 2.5 years before approval.246
Importantly, critics charge that the private compendia on which federal and state governments rely are too quick to certify off-label uses. A study of how those compendia addressed 14 off-label indications of anti-cancer drugs concluded that the compendia’s standards were lax: “The compendia’s stated methods varied greatly from their actual practices. Compendia cited little of the available evidence, often neither the most recent nor that of highest methodological quality.”247 And yet, there appears to be a durable political consensus that government health programs and private insurers must rely on these alternative sources of efficacy certification.
If most Americans are not aware of alternative forms of drug safety and efficacy certification, it is because the FDA crowds out private certification efforts. An author writing for the FDA even acknowledges, “Increasingly, responsibility for testing standards previously established as voluntary by the American Medical Association’s (AMA) Council on Drugs, the U.S. Pharmacopeia, and the National Formulary were taken up by the FDA” following the 1962 Kefauver-Harris Amendments.248 Indeed, for new drugs, the Kefauver-Harris Amendments effectively gave the FDA a monopoly over efficacy certification. The fact that the FDA holds this monopoly might account for the high cost of the FDA’s provision of this service.
Without the FDA’s monopoly on initial certification of safety and efficacy, alternative standards would likely be more numerous and more efficient. In addition to the informal certification mechanisms that already exist, medical societies might again enter the market by conducting safety and efficacy certifications for drugs used within their specialties. Health plans would face strong incentives to fund certification bodies and, through their coverage decisions, could educate enrollees about which drugs are ineffective, effective, and cost-effective. Integrated, prepaid group plans such as Kaiser Permanente would have a decided advantage in this market. The prepayment model gives such plans a greater incentive than other health plans to fund research that separates effective from ineffective drugs, and a fully integrated delivery system gives them the capability to conduct safety and efficacy studies themselves. To date, we have seen only a tiny glimpse of the voluntary drug certification activities a competitive market would provide.
In a market where the federal government respects the right to self-medicate, the problems of drug lag and drug loss would disappear. Unnecessary regulation would no longer delay consumers’ ability to access drugs. Consumers could use drugs when their assessment of a drug’s benefits and costs—and of the quality of the evidence of its benefits and costs—leads them to conclude that a drug is likely to benefit them.
As with eliminating government-imposed prescription requirements, eliminating premarket approval requirements would increase health literacy. Like a government-mandated prescription from a physician, an FDA approval can lead consumers to make less careful drug-consumption decisions than they would in its absence. A competitive market for drug certification, where different certifiers reach different conclusions about the same drug, would educate consumers that all drugs carry risks and that safety and efficacy are not binary concepts. A competitive certification system might develop different gradations of approval—much like the U.S. Preventive Services Task Force rates preventive services on a scale of “A” to “I”—that educate consumers about both the known effects of a drug and how certain they can be of those effects.
The FDA has even recognized the wisdom of using such gradations. When commissioning efficacy reviews of drugs approved prior to the Kefauver-Harris Amendments, the FDA allowed reviewers to assign drugs to different categories based on the strength of the evidence:
Their ratings on each claim for a drug fell into six categories: effective; probably effective; possibly effective, ineffective, effective but, and ineffective as a fixed combination (combination drugs for which there was no substantial reason to believe that each ingredient adds to the effectiveness of the combination).249
A competitive market for safety and efficacy certification would allow private certifiers to develop and use multiple, graduated categories that are better at educating consumers and clinicians about the benefits and costs of drugs, the strengths and weaknesses of the evidence for those benefits and costs, and the nature of drugs, health, and risk.
Respecting self-medication rights requires eliminating government premarket approval requirements for new drugs and for new information about existing drugs. If complete elimination of those requirements is not yet politically feasible, at a minimum Congress should eliminate the FDA’s ability to restrict truthful speech by manufacturers about their products and allow American consumers to purchase drugs that have won approval from designated certification agencies, including foreign regulatory bodies. According to one study, recognizing drug approvals by regulatory bodies in Canada and Europe between 2000 and 2010 would have given U.S. consumers quicker access to 37 “novel” drugs for which “no other FDA-approved prescription medicine had the same mechanism of action,” including 10 drugs treating mostly orphan diseases “for which no alternative therapy was available in the USA.” Such recognition would have allowed U.S. consumers to access those drugs a median of 13.6 months earlier.250
Intermediate Reforms
Congress and state legislatures can also take other intermediate steps that would expand consumers’ freedom to self-medicate, educate and acclimate them to making more of their medication decisions, and build support for eliminating government-imposed prescription requirements.
Automatic OTC Switches
Peltzman offers a proposal that in theory could accelerate the process of prescription-to-OTC switches and reduce the burden of government-imposed prescription requirements. Under the current system, manufacturers must prove to the FDA’s satisfaction that a drug is safe enough to sell directly to consumers, and the FDA decides what “safe enough to sell directly to consumers” means. Peltzman suggests that Congress create an objective standard of safety that, in effect, would remove the FDA from the decision of whether a drug is safe enough to sell OTC. He writes:
Why should the FDA wait for someone else to initiate this process? The FDA claims competence to decide when an adequate consumer label can be written. I suggest the FDA should periodically review existing drugs for eligibility for OTC sales. I further suggest that when any prescription drug passes certain milestones—x million prescriptions sold over y years with a risk profile similar to, say, ibuprofen or aspirin—there should be a rebuttable presumption that the drug becomes OTC-eligible. It would then be up to the drug’s producer or producers to take advantage of the opportunity.251
Once a manufacturer believes a drug has met that standard, it could sell the drug OTC without seeking the agency’s permission. The burden of proof would shift to the FDA to demonstrate why the manufacturer should not be able to sell the drug OTC.
Such a rule would still infringe on consumers’ freedom to make their own medical decisions, albeit less than the current system does. It would also face the challenge of determining objective criteria (“milestones”) that a drug must meet to free manufacturers and consumers from a government-imposed prescription requirement. Moreover, manufacturers of on-patent drugs would be unlikely to avail themselves of this option if there is no opportunity for competitors to offer OTC versions of the same drug.
Intermediate Classifications of Drug Access
Another option would be for Congress to create intermediate classifications of drug access between prescription-only and OTC. Congress could allow manufacturers to petition the FDA to designate drugs as “BTC,” “pharmacy,” “drugstore,” or “OTC/pharmacist,” under rules such as those in Table 1. Manufacturers could request an initial designation, and Peltzman-style rules could automatically move drugs to less-restrictive classifications either when they meet specified criteria or when select countries switch them to more-permissive categories. The FDA commissioner could order—or manufacturers, consumers, and others could petition—the agency to switch drugs to more-permissive categories even earlier.252
Creating less-restrictive government-imposed barriers to access would not fully respect the right of consumers to make their medical decisions. These barriers would still require consumers to obtain permission from government-appointed gatekeepers or otherwise restrict access. But they would eliminate many unnecessary doctors’ visits and their associated costs.
A flaw common to Peltzman’s OTC rule and government-created and ‑administered intermediate categories of drug access is that both still rely on the FDA to reclassify drugs. They rely on the agency either to lower the barriers it places in the way of consumers who want to purchase drugs or not to use its other powers to prevent Congress from lowering those barriers. The snail’s pace at which the FDA has considered reclassifying emergency contraception, oral contraception, and naloxone do not inspire confidence that the former approach would materially expand consumers’ freedom to self-medicate. The history of the FDA—in particular the agency’s use of its control of drug labeling to create a de facto yet never authorized power to classify drugs as prescription-only—suggests that the agency would defeat the latter approach as well.
Congress could try to circumvent this difficulty by adopting a rule that automatically assigns drugs to less-restrictive categories whenever any of a group of designated countries does so.
States Can Restore the Right to Self-Medicate
States can protect the freedom to self-medicate even if Congress and the FDA will not. The FDCA states that pharmacies may sell drugs that the FDA designates as prescription-only “upon a … prescription of a practitioner licensed by law to administer such drug.”253 Since states license medical practitioners, states have near-plenary authority to restore every individual’s right to self-medicate. If a state legislature wanted to do so, it could automatically license all adults in the state to administer all prescription drugs. In that event, all consumers would have to do to purchase any drug in a pharmacy is create their own prescription pads and present a self-administered prescription to a pharmacist.
States have taken some small steps toward making drugs more accessible. For instance, to make naloxone more widely available, all 50 states and the District of Columbia have found ways around the drug’s prescription-only classification. In most cases, a state’s chief medical officer issues a “standing order” to pharmacists to dispense the drug to any consumer who approaches the pharmacy counter.254 Other states empower pharmacists to be the prescribing practitioner. These reforms effectively make naloxone a BTC drug.
Though these reforms free adults to purchase naloxone essentially without a prescription, they remain inadequate. Experience has shown that the inconvenience of BTC access combined with reluctance to reveal government-stigmatized opioid use prevents many opioid users, their friends, and family members from availing themselves of the drug. Numerous reports of pharmacists refusing to participate in naloxone distribution—some because they believe they are enabling a dangerous drug habit—likewise obstruct the wider distribution and use of naloxone.255 This problem illustrates that BTC status is inadequate and that manufacturers should be free to sell the drug OTC, including in vending machines.
With regard to oral contraception, California, Colorado, the District of Columbia, Hawaii, Idaho, Maryland, New Hampshire, New Mexico, Ohio, Utah, Oregon, Tennessee, Washington, and West Virginia let consumers obtain oral contraceptives directly from a pharmacist, with varying restrictions.256 Unfortunately, those states have implemented this approach with uneven success.257 And even if successful, BTC status still fails to respect the right of consumers to self-medicate. The ACOG writes,
Pharmacist prescribing laws are not the same thing as over-the-counter access. Requiring a pharmacist to prescribe and dispense oral contraceptives only replaces one barrier—a physician’s prescription—with another. This is not going to allow us to reach women who remained underserved by the current prescribing requirements.…
We know from evidence and experience that oral contraceptives are safe enough for over-the-counter access, and do not require any prescription at all.258
BTC access is inadequate and no substitute for respecting the right of individual consumers to self-medicate.
Civil Disobedience
The laws that allow the FDA to block drugs from the market and to force consumers to obtain prescriptions from government-approved gatekeepers are harmful, unjust, and immoral. They prevent consumers from exercising their right to self-medicate and prevent manufacturers both from selling safe, effective, and even life-saving drugs directly to consumers and from providing consumers truthful information about drugs.
Trailer for the 2013 film Dallas Buyers Club portrays civil disobedience by HIV patients at the height of the AIDS epidemic in 1985.
The hardships that pharmaceutical regulations impose on patients are so great that disobedience to such regulations is common—and in many cases receives tacit or explicit government sanction. The 2013 Contraception survey found that in some 56 countries, pharmacies routinely sell oral contraceptives without a prescription even though the law forbids it.259 Other examples of disobedience to pharmaceutical regulations include buyers’ clubs formed by AIDS patients, patients importing lower-priced and/or unapproved drugs from foreign countries (often at the behest and with the assistance of state governments), and state governments licensing medical marijuana dispensaries.260
Resistance to Reform
Reform will not come easily. Many well-funded interests, inside and outside of government, will oppose restoring the right of individuals to self-medicate. Manufacturers who fear price competition might oppose reform because it would mean a larger share of their customers would be price-sensitive rather than relatively price-insensitive consumers, insurance companies, and government programs. Manufacturers who classify a drug as prescription-only and charge prescription prices might see a threat from competitors marketing the same drug OTC. Recall how a manufacturer of naloxone responded to a Capitol Hill briefing about the need to switch naloxone to OTC.
Even though a significant number of drugs, especially those for complex and highly specialized problems, would remain prescription-only, the medical and dental professions may oppose reform because it would reduce the number of office appointments and the control those professions hold over the medical decisions of adults.
The greatest resistance may come from within government itself. Officials in Congress and the FDA will argue that it would be unwise and unsafe to divest them of the authority to make medical decisions for others. In part, government officials’ opposition to the right to medical self-determination springs from the fact that they have never seen and cannot imagine the patient-safety and patient-education efforts a competitive pharmaceutical market would spur innovators to create.
Conclusion
Government-imposed prescription requirements create unnecessary barriers to beneficial drugs, including by encouraging excessive drug prices. The evidence that suggests such requirements harm rather than help consumers becomes more plausible when one considers how both physicians and government have steered consumers toward hazardous drugs.
With many consumers struggling to afford the medications they need, the need for reform is urgent. After the FDA rejected a manufacturer’s petition to make emergency contraception available OTC in 2004, the ACOG wrote, “Any delay in OTC status … means that every day teenagers and women have difficulty obtaining emergency contraception.”261 The same is true for routine-use oral contraceptives, life-saving naloxone, and other safe and effective medications.
Beyond economic considerations, government-imposed prescription requirements and premarket approval requirements deny consumers their right to make their own medical decisions, as surely as if government allowed doctors to lie to consumers or perform unwanted medical procedures on them. To preserve the FDA’s power to require prescriptions is to subject consumers to the sort of paternalism that patient advocates have fought for decades to eliminate via the doctrine of informed consent. A regulatory system under which “a 12-year-old can purchase a lethal dose of acetaminophen in any pharmacy for about $11, no questions asked” but that forces competent adults to obtain permission from a government-appointed gatekeeper before purchasing nonlethal oral contraceptives or life-saving naloxone is not just perplexing or inefficient; it fails to respect the fundamental and equal dignity of those consumers.262
Eliminating the FDA’s authority to impose prescription requirements on drug manufacturers and consumers would provide a safe and effective way to bring beneficial medications to patients who need them. Peltzman writes:
With the pressure mounting for action to restrain drug prices, you might think that speeding up and broadening the OTC transition would be on the FDA’s priority list or the priority list of its critics. But the topic is little discussed by anyone. This neglected area deserves more scrutiny.… Moving more drugs to OTC status is no free lunch, but it is as close to one as consumers are likely to get in the health care sector.263
The knowledge that some individuals would inevitably self-medicate in ways that harm their health should not delay reform. Americans do the equivalent every day when they exercise their right to refuse potentially beneficial medical treatments.
The utilitarian argument for eliminating government-imposed prescription requirements, moreover, is not that manufacturers and consumers of pharmaceuticals will never make harmful mistakes. It is that government is right now making far more harmful mistakes than would a free consumer populace aided by price competition, quality competition, third-party certification, innovation, greater health literacy, and the threat of tort liability.
In any event, the burden of proof lies not with those who seek to restore the right of individuals to make their own medical decisions. It lies with those who would preserve laws that allow government to interfere in personal medical decisions. The individual-rights argument for eliminating government-imposed prescription requirements is that government has no legitimate authority to interfere in the first place.

About the Authors



This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.